Mogul in Action: How Conformations Influence Macroscopic Properties

Investigating the Relationship Between Conformation and Macroscopic Properties in Isoflavones
Here we highlight a paper by Eric Sperlich and co-workers, where the team studied the solid-state structures of three very similar isoflavones to investigate how supramolecular packing and conformations influence their macroscopic properties.
The full article can be accessed here – CrystEngComm, 2022, 24, 4731-4739.
What Are Isoflavones?
Isoflavones are natural products found in plants that have attracted attention for their bioactivity and ability to bind to the human estrogen receptor (hERα).
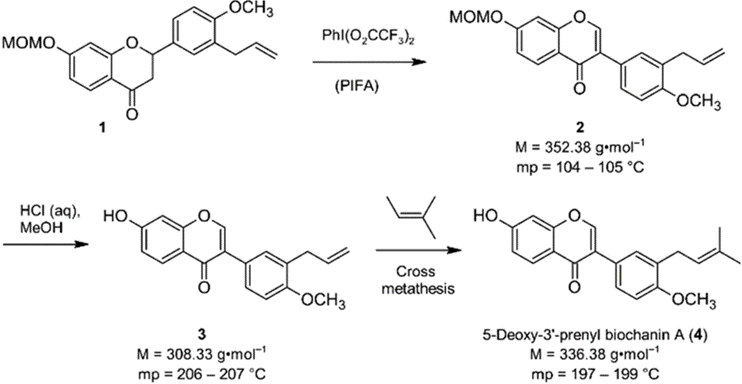
In this work, scientists synthesized isoflavone 5-deoxy-3′-prenylbiochanin A (4) to study the anti-infective activities of metabolites from E. sacleuxii to bacterial pathogens.
Macroscopic Properties
Solubility is a key parameter for pharmaceutical molecules as the bioavailability of a drug is strongly dependent to its dissolution rate.
Interestingly, different solubilities were seen for compound 4 and the precursors compound 2 and 3, alongside other macroscopic properties like their melting point.
A Geometry Investigation
The group analysed the solid-state structures of 2, 3, and 4, aiming to find the relationship between their supramolecular structures and macroscopic properties.
Using Mogul, the team were able to investigate the geometries of the compounds and compare these with structurally related compounds from the Cambridge Structural Database (CSD).
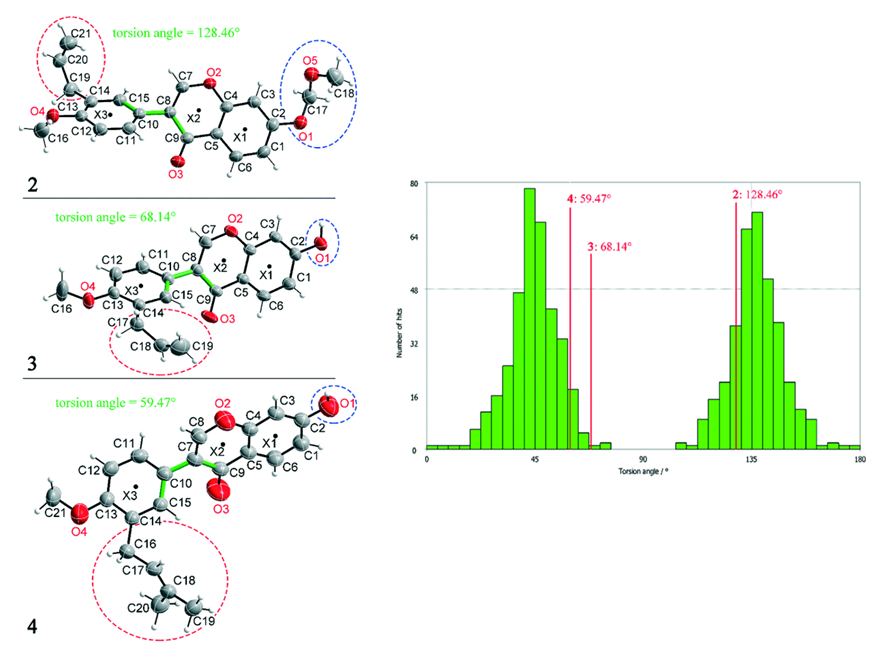
Investigating the Macroscopic Properties
Synthesis and Properties
This work reports the synthesis of the isoflavone 5-deoxy-3′-prenylbiochanin A (4).
The team observed that that despite having a similar molecular structure, compound 4 and the precursors compound 2 and 3 crystallized in different space groups, and exhibited different lattice energies, meting points, and solubilities.
To rationalize these findings, the team analysed the geometric conformation parameters and the intermolecular non-covalent interactions for compounds 2, 3, and 4. Using Mogul the geometry of the compounds was validated and compared with 640 structurally related compounds found in the CSD.
Analysing the Solid-State
It was seen that compound 3 presents an unusual torsion angle of 68.14° (with the ideal one being 45°) between the bonds highlighted in green in the figure above, and that only three compounds out of 640 had such a small angle.
The scientists hypothesized that the torsion angle could be dictated by intermolecular interactions, as the two aromatic rings, in this case an aryl group and a benzopyran group, can be independently influenced by these.
Hence, they tested the assumption that a large deviation from the expected torsion angle was correlated to the formation of stronger intermolecular interactions.
The Intermolecular Interactions
The hydrogen bonds and stacking interactions in the structures were hence studied performing Hirshfeld surface analysis and using the CCDC’s Aromatics Analyser. The analysis revealed the physicochemical properties of compounds 2, 3, and 4:
- The low lattice energy and melting point of 2 were attributed to the absence of a strong intermolecular O-H···O hydrogen bonds, and to the repulsion between the bulky ring substituents that prevents the formation of stacking interactions. The presence of non-polar groups, added to the lack of intermolecular interactions, also explains its good solubility in organic solvents.
- Compounds 3 and 4 present instead strong one-dimensional O-H···O hydrogen bonds and strong parallel-displaced stacking interactions between the rings, which explain their higher melting points when compared to compound 2. The presence of the OH group at C2 is also responsible for the lower solubilities in nonpolar solvents.
Link to Macroscopic Properties
Geometry analysis showed that when only weak hydrogen and stacking interactions are present in the structure (compound 2), the rings are not locked in a specific orientation, hence exhibiting a torsion angle with a small deviation from the ideal one.
When instead the benzopyran ring is fixed by both strong hydrogen and stacking interactions, which is the case in compounds 3 and 4, the torsion angle is strongly influenced by the intermolecular interactions on the aryl ring. This leads to a greater deviation from the ideal torsion angle for compounds 3 and 4 when compared to compound 2.
It was hence observed that a greater deviation from the ideal torsion angle is seen when there is an increase in number and strength of intermolecular interactions. This trend is also followed when considering the macroscopic properties of compounds 2, 3, and 4, with stronger interactions leading to increased lattice energies and melting point, and decreased solubilities.
Features of Mogul
What is Mogul?
Mogul is a tool that is used by academic researchers and industry leaders to validate the geometry of experimental or predicted structures.
Mining the entire CSD, Mogul identifies intra and intermolecular geometry preferences, and predicts the likely chemical conformations.
A Geometry Analysis
Mogul is easily accessible and can be launched directly from Mercury. It can be used to quickly identify unusual features in a structure, performing a general geometry analysis or a more targeted feature search depending on the research goals.
Mogul can also display the distribution of bond lengths, angles, ring geometries, and torsions of the expertly curated structures in the CSD.
Validating Crystal Structures
Watch our video “How to validate atom-type assignment using Mogul” to learn how to validate a crystal structure refinement using Mogul, and follow our free online training course “Analysing molecular geometries 101 — basics of Mogul” to find out more.