Aromatics analyser: your questions answered
The Aromatics Analyser is a new feature available in Mercury as part of the CSD-Materials and CSD-Enterprise suites. It was launched with our last release 2020.1 in April. Our users have been asking questions during our virtual events, as the What’s Up customer webinar, and we decided to share with you some of the most common questions.
How does the stability of the aromatics interactions relate to the overall stability of the crystal structure?
There will be cases where the aromatics interactions are aligned with the overall crystal structure stability, however, this will not always be the case since all the interactions in the structure contribute to the overall stability such as H-bond interactions, weak interactions, repulsive interactions, etc.
Is there any dispersion interaction included?
The model was trained on benzene dimers only, hence dispersion interactions were not considered as neighbours. However, we did test rings that came from dispersing interactions to check whether we cover this type of geometry and we do. Please see the picture below as an example. The DFT and Neural Network energies of the highlight rings extracted from QINCUL01 refcode are very similar.
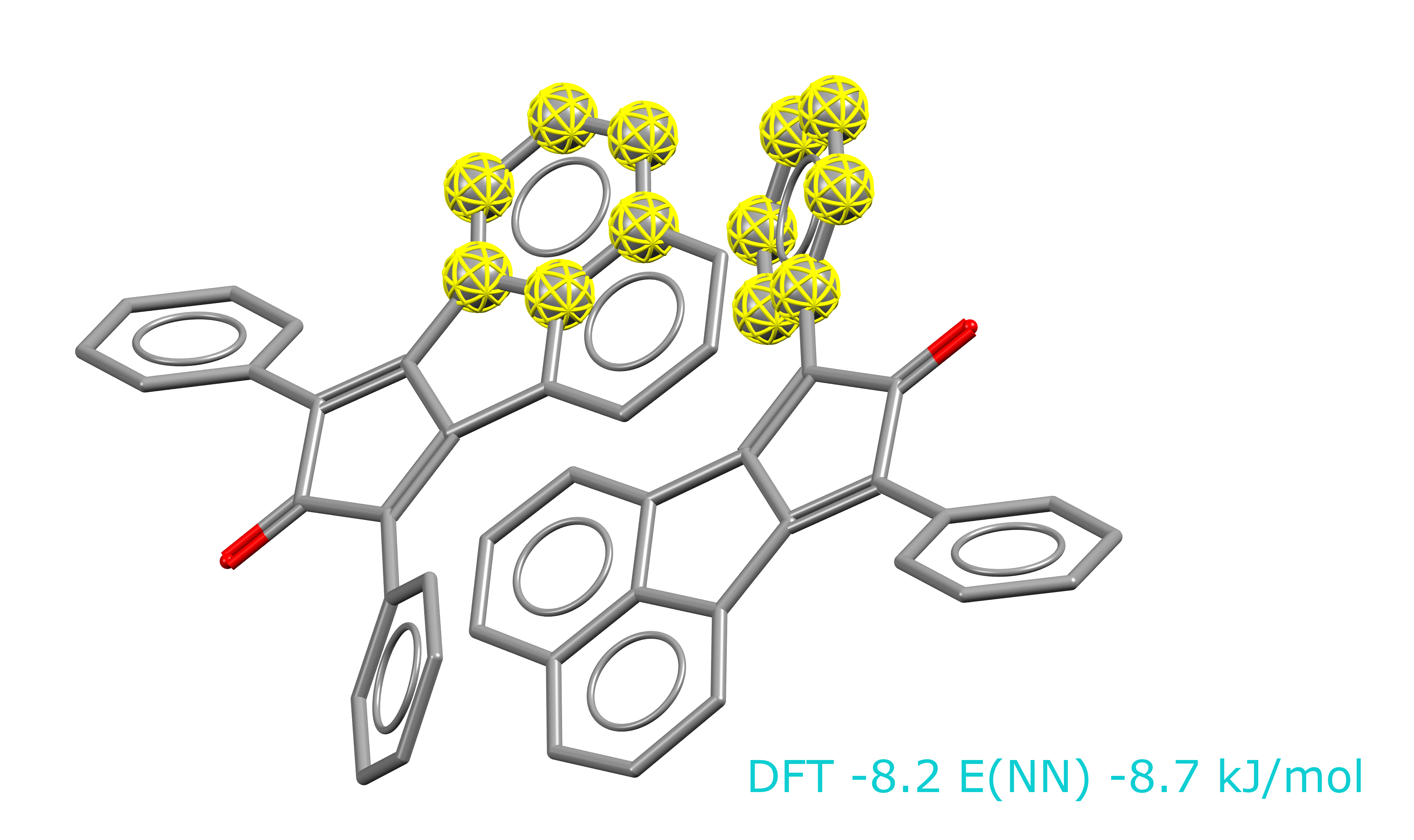
Were the DFT calculation corrected for dispersion?
Yes, the calculations were corrected for dispersion, we used D3 Grimme dispersion correction.
Can I use Aromatic Analyser for other aromatic rings than benzene – for example imidazole, indole, Cp, porphyrins, pyridines?
The model was trained on benzene dimers only, hence any heteroatoms or five members rings are not available in the current implementation. Therefore, the tool will simply not perform any scores or show any information in the displayed windows of the Aromatics Analyser on this type of rings.
To deal with other types of aromatic interactions, can we transfer the Neural Network model to heteroatom systems based on a few energy calculations?
This needs to be investigated by calculating a few points in the potential energy curve for hetero atom rings and compare the trend with benzene interactions. The geometries and the centroid distances should also be considered.
In structures there is a balance between attractive and repulsive interactions, are all interactions in the training set attractive?
The training set only included the benzene dimers interactions that were negative in energy (overall attractive). Any repulsive interactions between the dimers will be captured in the overall energy outcome, and if the benzene rings are very close and overall repulsive the score will be 0.
To find out more about Aromatics Analyser you can watch the May What’s Up webinar on-demand here, follow the self-guided workshop here , or watch our 3 minute video on How to use Aromatics Analyser here.
Contact us here if you don’t currently have access but want to try this feature.