Q&A with Mihaela D. Smilova: building scalable drug discovery processes with fragment-based drug design
In this Q&A-style blog with Mihaela D. Smilova (postgraduate researcher at the Centre for Medicines Discovery at Oxford University), learn about some of the advantages of fragment-based drug design, which she recently used for a drug design methodology called “ensemble hotspot mapping.”

We recently featured the paper “Fragment Hotspot Mapping to Identify Selectivity-Determining Regions between Related Proteins,” co-authored by Smilova and researchers at Oxford, Exscientia, and CCDC. In the paper, the authors show how ensemble hotspot maps can automate identifying key structural differences that contribute to the selectivity of a compound for one protein over another.
The benefits and scalability of fragment-based drug design
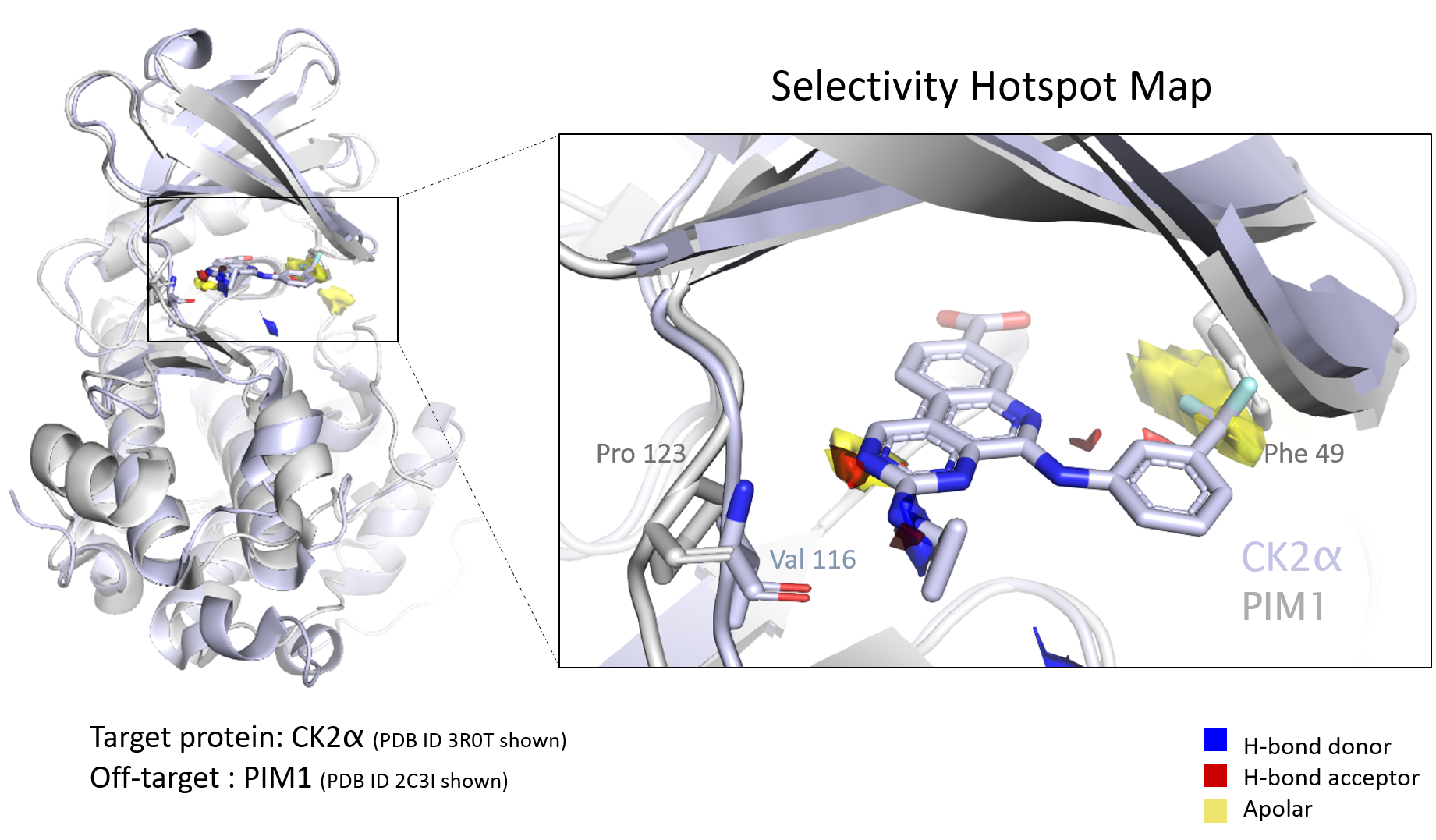
Ensemble hotspot mapping leverages fragment-based drug design, which is becoming an increasingly common way to progress through early-phase drug discovery. [1,2] We asked Smilova about the important role of fragment-based drug design in hotspot mapping, and some of its general advantages in target identification and hit-to-lead efforts, like assessing pocket druggability. Here’s what she had to say.
The paper notes that fragment hits may provide a more efficient exploration of the relevant chemical spaces as compared to larger compounds commonly used in high-throughput screens. Can you explain a bit more?
Fragments require smaller screening libraries while getting better coverage of chemical space. Chemical space is estimated to increase eight-fold for every heavy atom added to a molecule.[3] This means that the fragment space is many orders of magnitude smaller than the drug-like space—allowing the use of much smaller screening libraries.
What effect does this have on hit rates?
Fragment-sized molecules have higher hit rates. As the complexity of the molecules decreases, the chance of finding a complementary region of the binding site increases. The trade-off is that smaller compounds bind more weakly, requiring more sensitive biophysical methods. However, the higher hit rates mean that starting chemical matter can be obtained with a greatly reduced number of experiments.
What are the advantages of using smaller fragments from a design perspective?
Fragments have good solubility and leave space for optimization into high-quality leads. Less sensitive methods require larger compounds to be used for screening. These compounds may make suboptimal interactions with the binding site, and their potency may be driven by lipophilic interactions, which can cause problems with permeability, metabolism, and toxicity downstream in the discovery campaign. Fragments provide more polar and soluble starting points, making few, but key interactions with the binding site.
Learn more
Read the full press release highlighting how ensemble hotspot mapping can automate hit-to-lead research.
Download the whitepaper with further details on ensemble hotspot maps and their research impact.
Read the paper, “Fragment Hotspot Mapping to Identify Selectivity-Determining Regions between Related Proteins.”
View the CCDC CSD Python GitHub repository, where you can find the scripts to automate common tasks in structure-based drug discovery.
[1] Erlanson, D.A., et al. “Twenty years on: the impact of fragments on drug discovery,” Nat. Rev. Drug Discovery. 2016, 15, 605–619.
[2] Taylor, S.J., et al. “Discovery of Potent, Selective Chymase Inhibitors via Fragment Linking Strategies.” J. Med. Chem. 2013, 56, 11, 4465–4481
[3] Hall, R. J., et al. “Efficient exploration of chemical space by fragment-based screening,” Progress, Biophysics and Molecular Biology. 2014, 116(2), 82–91.