Why do aromatic interactions matter?
In our 2020.1 CSD Release, we launched the Aromatics Analyser – our first feature in Mercury based on a Neural Network that leverages deep learning. The tool allows you to quantitatively assess aromatic ring interactions and their likely contribution to the stability of a crystal structure. In this blog, let’s explore why it’s so important to understand aromatic interactions using ibuprofen and benzoic acids as examples.
What are aromatic-aromatic interactions?
Aromatic rings are highly stable due to the arrangement of the π-electrons situated above and below the plane of the aromatic ring, which form a π-electron cloud. The π-electrons of these planar compounds are free to cycle around the circular arrangements of atoms found in the aromatic moieties. This stems from the resonance found in planar ring systems, like benzene. The flat faces of aromatic rings also have partial negative charges due to the π-electrons.1 Similar to other non-covalent interactions – including hydrogen bonds, electrostatic interactions and Van der Waals interactions – aromatic interactions can greatly affect the stability and interactions of a crystal. They are the strongest such interactions after hydrogen bonding.
Hydrogen Bonds > Aromatic Interaction > Van der Waals
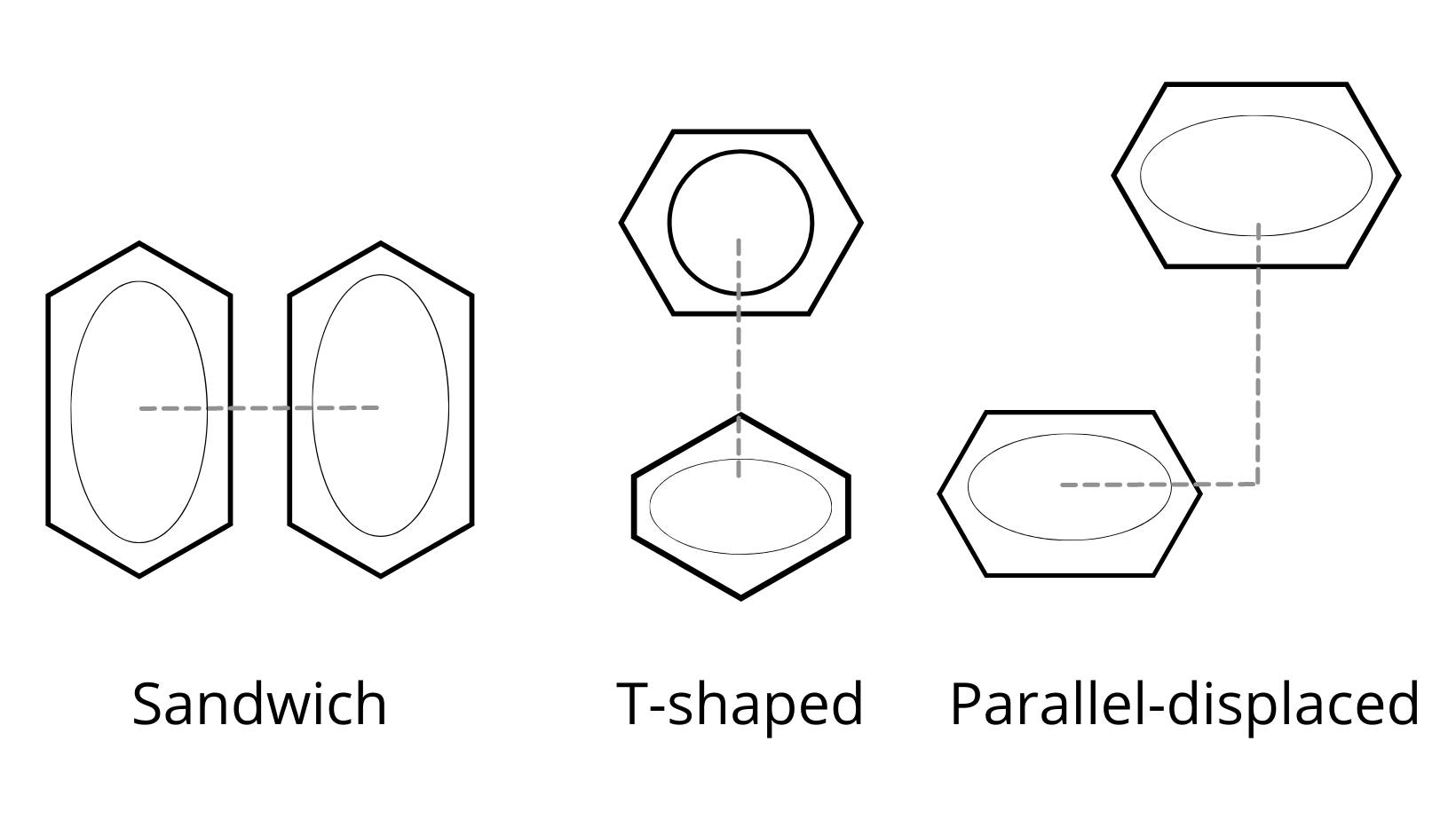
The strength of the aromatic interaction relates to the orientation of the benzene dimers, and there are a few possibilities observed in a crystal structure, as indicated in the image above.
Aromatics and polymorphism
In previous blogs, we’ve discussed how to use CCDC’s Hydrogen Bond Propensity (HBP) tool to manage risks associated with polymorphs. For example, the output of the tool includes:
- HBP chart that maps the mean H-bond propensity vs. mean H-bond Coordination. (The closer a structure appears to the lower-right corner of the HBP chart, the more likely the H-bond network.)
- HBP Score Table that ranks the likelihood that an H-bond network will form. (The most likely H-bond network will score the highest propensity and appear first on the table.)
But what if a system doesn’t have H-bonding? Or what if H-bonding networks are the same in two different polymorphs? For example, in the image below two polymorphs of ibuprofen show just one point on the HBP map.
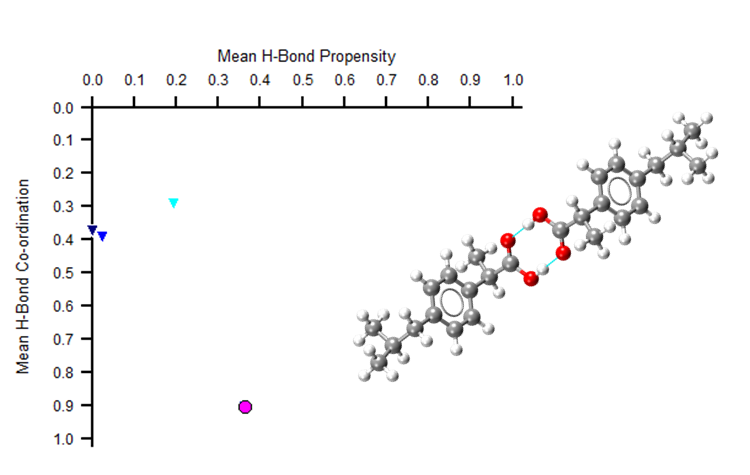
Both polymorphs appear as the pink circle above. Meaning that the second polymorphic form could be missed. This highlights how an HBP analysis alone may not fully characterise your molecule. Additionally assessing the aromatic interactions can help distinguish between such polymorphs. Read more in this blog on how to use the aromatic analyser tool.
Aromatic interactions and solvates
Solvent choice and supersaturation conditions can yield different crystal forms. Depending on the polymorph present, different non-covalent interactions can dominate. For example, in the paper Polymorph Selective Role of Hydrogen Bonding and π−π Stacking in p‑Aminobenzoic Acid Solutions, Bobrovs et al.2 combined solution NMR spectroscopy and molecular dynamics (MD) simulations to understand the structural and dynamical features of p-aminobenzoic acid (pABA, CSD: AMBNAC) in solution and pABA crystals nucleation.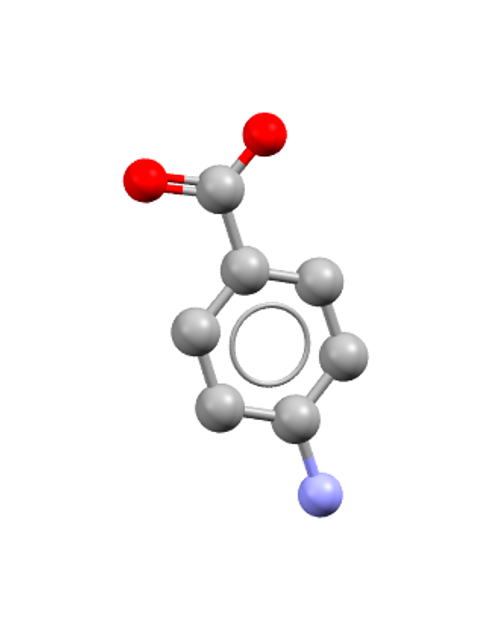
AMBNAC: p-Aminobenzoic acid
The researchers’ NMR results showed that the dominant interactions stabilizing pABA oligomers are solvent-dependent. In organic solvents, hydrogen bonds dominate, while water promotes π−π stacking. Their NMR experiments provided a clear indication that different intermolecular interactions drive self-association in the different solvents, which their MD simulations supported.
Non-polar interactions and nucleation rates
Crystallization from solution is vitally important to pharmaceutical, agrochemical and speciality chemical industries for purifying and creating products in solid forms; however, the relationship between the kinetic process of nucleation and the molecular and crystal structures of a crystallizing solute can remain unclear.3 In their paper, Can molecular flexibility control crystallization? The case of para-substituted benzoic acids, Tang et al. looked at the relationship between molecular conformation and the ease of crystallization. They investigated the relative importance of solution chemistry, solid-state interactions and conformational flexibility in crystallization – confirming the important role of intermolecular stacking interactions in determining relative nucleation rates. They concluded:
- Increased conformational flexibility (i.e., a higher number of rotatable bonds) does not, on its own, control the relative rates of nucleation.
- Of the studied series of para-substituted benzoic acids, other solid-state features – notably continuous dispersive and stacking interactions – dominate nucleation rates and the ability to crystalize.
Designing crystal growth modifiers for non-polar crystals
Better understanding how additives can modify and control the crystallization of desired solutes has several applications regarding habit modification, inhibition of crystal growth and the control of polymorphic forms. These efforts have given rise to the term “tailor-made additives.”4 In the case of polar molecules, such research often focuses on directional inter-molecular connections, like H-bonding networks. But with weakly polar molecules or in the absence of H-bonds, another approach is needed. In their paper Concerning the selection of crystallization modifiers for non-hydrogen bonded systems: the case of benzophenone, Black et al. outline a design strategy for the selection of crystal growth modifiers for non-polar crystals using benzophenone as the example.
- The strongest intermolecular interaction in the stable form of crystal benzophenone combines two strong phenyl-phenyl approaches.
- The researchers identified two additives that disrupt that interaction and in doing so modified the morphology and decreased the crystal growth rates of benzophenone crystals.
Learn more
Learn more about the neural network methods used to build the analyser.
Read the answers to common questions and more about the tool’s functionality.
Watch our webinar, “Behind the paper – Can solvated intermediates inform us about nucleation pathways” in which Dr Roger J. Davey (co-author on some of the work discussed here) walks through his team’s research.
1Bioinformation. 20128 (24), 1220–1224. (DOI: 10.6026/97320630081220)
2 Crystal Growth & Design 2021 21 (1), 436–448 (DOI: 10.1021/acs.cgd.0c01257)
3 Chem. Sci., 2021, 12, 993-1000 (DOI: 10.1039/D0SC05424K)
4 CrystEngComm, 2021, 23, 1281-1293 (DOI: 10.1039/D0CE01547D)