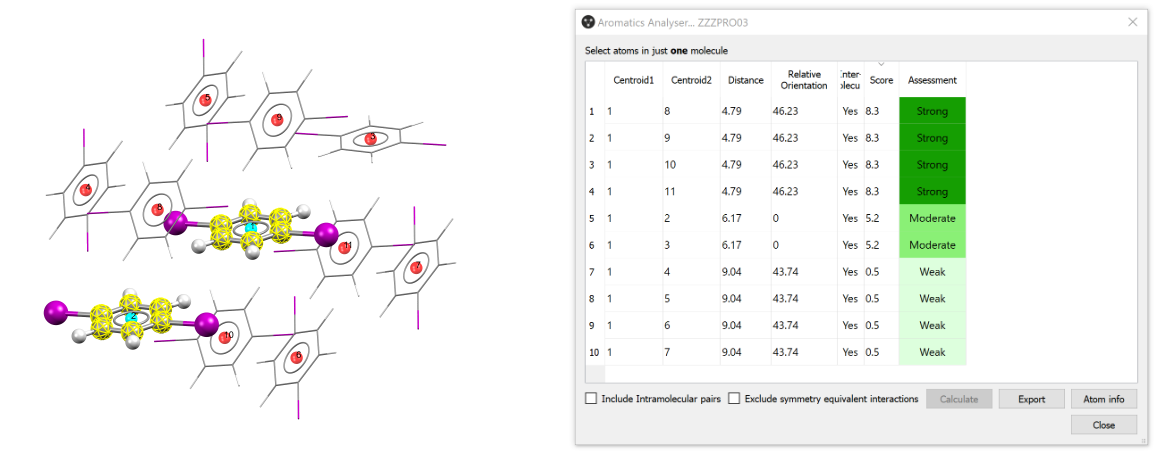
New for 2020.1 release – aromatics analyser in Mercury
We live in exciting times for Artificial Intelligence (AI) - with the rise of new and easy to implement Machine Learning (ML) algorithms. Many of us would sooner trust a GPS to take us from point A to point B than consult a map ourselves, and robots are already being used to perform medical procedures. But what do all of these advanced techniques and algorithms mean for us as scientists and how can we use them to advance science? Presumably, many would ask if AI approaches can help, or even replace scientific experiments?
We can definitely see this happening already, this year a new antibiotic (halicin) has been discovered using a Deep Learning Approach by predicting a range of potential drugs and therefore significantly reducing the number of experiments required to obtain a drug candidate compound (Stokes et al., 2020, Cell 180, 688–702). A number of ML approaches have been successfully utilised in the materials science and crystallography fields for predicting properties such as solvent formation (Xin et al., Cryst. Growth Des. 2019, 19, 1903−1911), crystallisation (Wicker et al., CrystEngComm, 2015,17, 1927 –1934) and crystallographic space groups (Liu et al., Acta Cryst. (2019). A75, 633–643). ML has been proved also accurate in replicating quantum mechanical calculations such as NMR spectra (Gerard et al., Chem. Sci., 2020, 11, 508–515).
In our 2020.1 CSD Release we are launching the Aromatics Analyser component, our first feature in Mercury to be based on a neural network, which allows you to quantitatively assess aromatic ring interactions. The model is based on a geometric description of aromatic interactions involving the position of two phenyl rings relative to each other and is based on a large number of quantum mechanical calculations. The outcome of this model is a score (from 0 to 10) indicating whether the aromatic interaction is weak (0-3), moderate (3-7) or strong (7-10).
This approach provides insight into aromatic interactions by quickly visualising them and picking out stabilising interactions. You can quickly and easily analyse the likely contribution of aromatic interactions to the stability of a crystal structure. It can be applied for instance to systems where H-bonding are absent or if H-bonding networks are the same in two polymorphs.
This feature provides quantitative assessment of each aromatic interaction in comparison to the best geometry that could be achieved for a phenyl to phenyl contact. This is displayed as a score from 0 (no stabilising contribution) to 10 (an ideal aromatic interaction geometry). The input structure for analysis is selected from the Mercury Structure Navigator, i.e. a CSD entry, in-house database entry, or a structure loaded into Mercury from a 3D structural file such as a CIF or MOL2.
Note that only six-membered aromatic rings will be considered by the Aromatics Analyser. The rows are interactive and allow the user to highlight a specific ring∙∙∙ring interaction in the 3D visualiser.
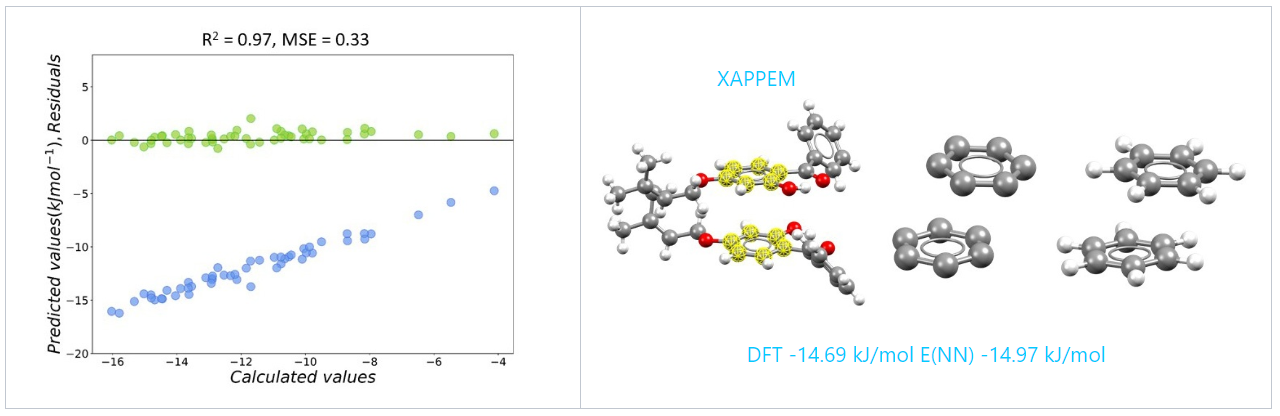
The neural network model reproduces the quantum mechanical calculations with 97% precision. To test the performance of our model we looked at real crystal structures containing aromatic functional groups from the CSD. To calculate the strength of aromatic interactions we took the following steps:
- the atomic positions of the phenyl dimers were extracted
- substituents were replaced with H atoms
- then DFT quantum mechanical calculations were performed for these dimers.
We compared the DFT with the neural network model outcome and, as you can see for refcode XAPPEM, the energy is very well reproduced by the neural network: DFT -14.69 kJ/mol E(NN) -14.97 kJ/mol.
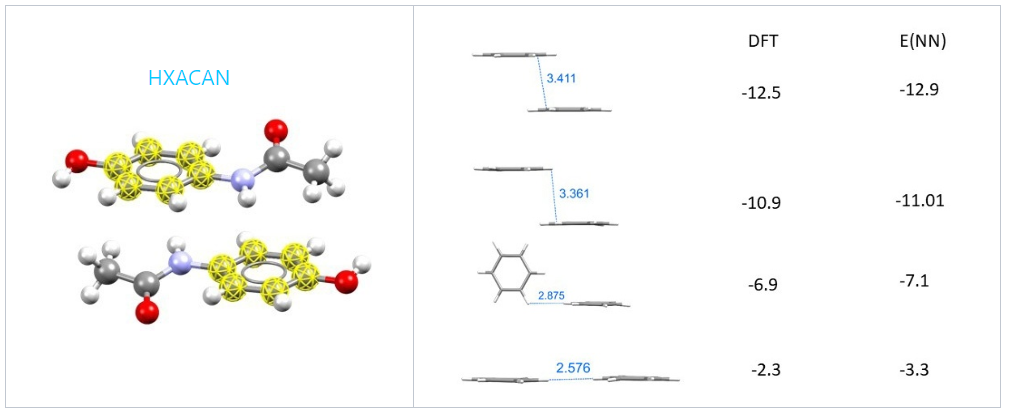
The neural network prediction also performed well over a heterogenous orientation of phenyl groups which were extracted from paracetamol (refcode HXACAN).
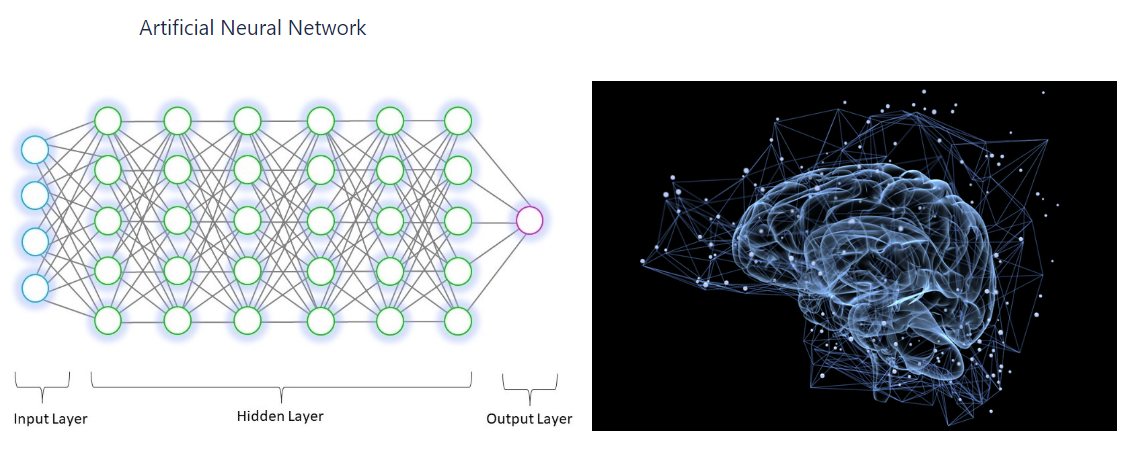
How neural networks function:
The input values are multiplied with initial random weight values (close to zero, but not zero). In our case the input values were the geometric parameters (e.g. atom-atom distance, centroid-centroid distance and plane-plane angle).
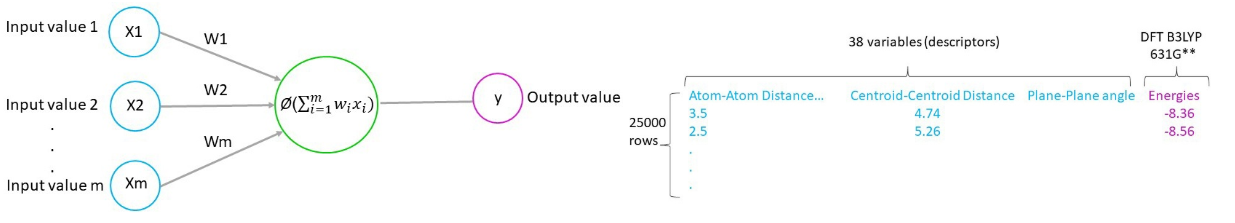
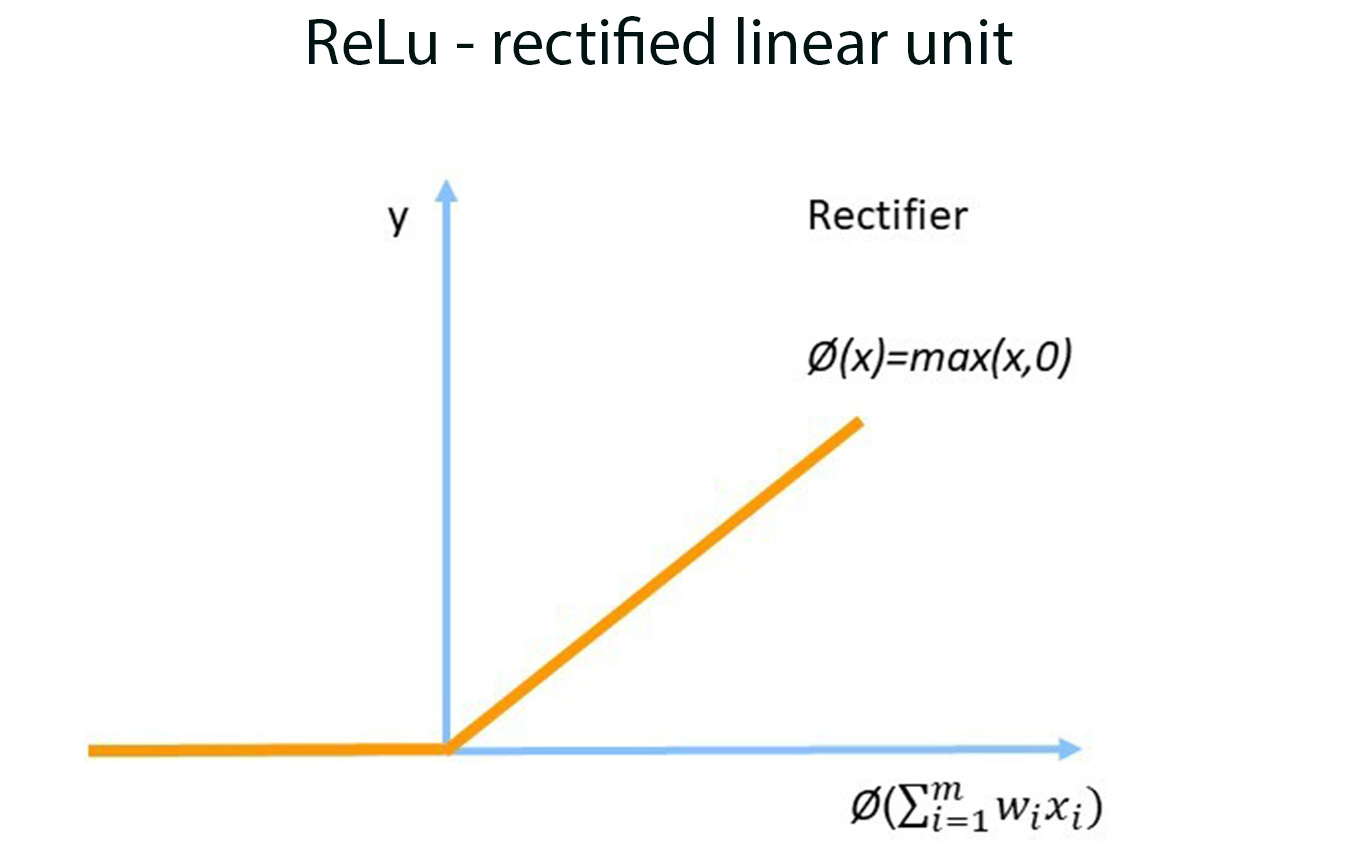
Each node in the neural network will use a function – in this case we selected the Rectified Linear Unit (ReLu) to produce an output value.
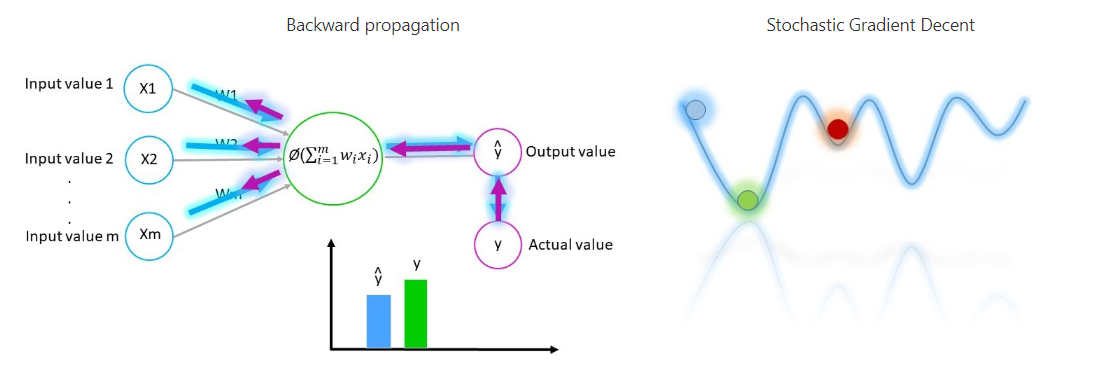
The output values is compared with the actual value and then something called “backwards propagation” happens where the weights are adjusted in such a way that minimises the difference between the output value and actual value. To minimise those values and to ensure that no local minimum occurs the algorithm uses something called Stochastic Gradient Descent.
If you are curious of how the world could look with fully functional AI mechanisms, or you want to teach your children more about neural networks, these two books are for you:

If you would like any more information about the Aromatics Analyser component, please don’t hesitate to get in touch, you can email us at .