Validation of Experimental Crystal Structures
This blog discusses the validation of crystal structures using tools to analyse both molecular geometry and intermolecular packing.
Introduction
Poor crystal structure refinements are generally caused by one of three main issues. First, the conditions under which the experiment was performed may have been less than ideal, e.g. room temperature or high pressure. Secondly, the refinement may be poor due to the limited accuracy of the data; this can be caused by weak diffraction, poor crystals and severe disorder, amongst other reasons. Finally, the structure may simply be wrong; important examples of mistakes include incorrect assignment of element types, missed symmetry in the structure and too few or too many hydrogen atoms.
For a given structure, it would be ideal to have the ability to quickly and confidently decide, for poor refinements, which of these categories the structure fits into. This would mean that one could decide efficiently whether a structure is acceptable, needs further refinement, or is simply incorrect. Structure validation tools are designed to provide help in the making of this decision.
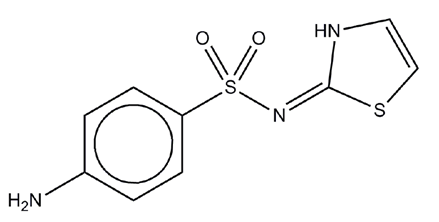
An example of a relatively poor quality crystal structure refinement can be seen for a high-pressure, single crystal, X-ray determination of the drug compound sulphathiazole (Figure 1) at 2.3 GPa (equivalent to 23 kbar). This structure determination has been performed at thousands of times atmospheric pressure (approximately 1 bar), in which region phase transitions are common, but the intramolecular geometry is unlikely to change.
The compound sulphathiazole is a short-acting sulpha drug used as an oral and topical antimicrobial treatment. Form I of sulphathiazole (determined by Kruger & Gafner1) is metastable at ambient conditions but has a lifetime that varies from days to years depending on defects in the crystals. This form is known2 to exhibit highly anisotropic thermal behaviour unlike the three lower melting polymorphs (forms II, III & IV). Sulphathiazole was studied at extreme pressures to determine whether the unusual behaviour with respect to temperature was mirrored by the behaviour of the compound with respect to pressure. Experimental procedures for X-ray structure determination at pressure followed the previous work of Dawson and co-workers.3
Method
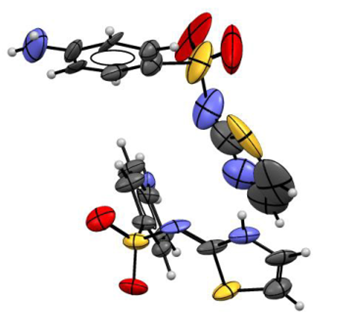
The refinement for the high pressure structure of sulphathiazole form I showed significantly anisotropic thermal parameters (Figure 2) as well as exhibiting relatively poor statistics for data quality, e.g. R-factor of 13.0% and residual peaks in the electron density of around +0.9 e/ų. These factors can potentially be indicators of an incorrect or incomplete structure, but could also be simply due to a lack of available data.
In order to ascertain whether there are any significant problems in the chemistry and crystallography for this refinement, it is important to compare the refined structure with other, related structures that have been previously observed. The program Mogul4 was used to analyse the intramolecular geometry parameters in the structure. This software package automatically retrieves relevant data for each parameter from the Cambridge Structural Database (CSD)5 and determines whether the parameters are unusual or not. The results include a statistical analysis of how commonly observed each geometrical parameter in the structure is, as well as a judgement of whether there is enough data to make an informed decision in each case.
Additionally, when validating a structure it is beneficial to examine the intermolecular interactions with the crystal packing pattern. These can be analysed again by using the knowledge base of related structures observed in the CSD – the frequency of occurrence and most likely geometry for the intermolecular contacts were investigated using the CSD-Materials module of Mercury6 and IsoStar7 respectively.
Results
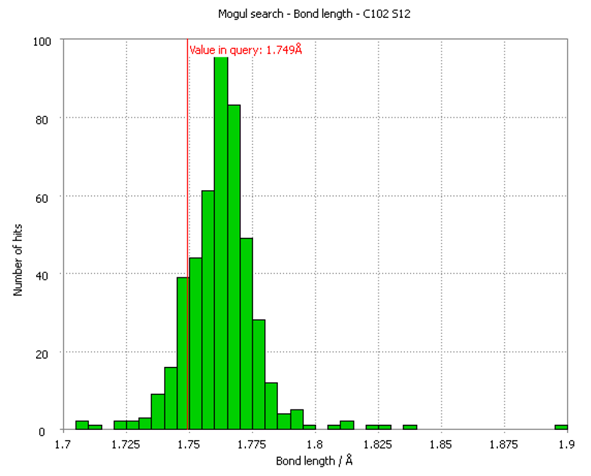
Due to the two crystallographically distinct molecules in sulphathiazole form I, there are 34 unique bond lengths, 48 bond angles, 22 torsion angles and 2 covalently-bonded rings in the structure. The Mogul histogram for the bond length with the highest Z-score (a statistical parameter indicating a normalised distance from the mean value) in the structure is shown in Figure 3. The observed bond length between atoms C102 and S102, highlighted as a red line in Figure 3, is still comfortably within the distribution of equivalent bond-lengths in the CSD and therefore cannot be considered unusual.
Similar results were found for the analysis of the bond angles, torsion angles and ring geometries in the structure, with the parameters deviating furthest from the observed data still residing within the observed CSD distributions. All of the intramolecular geometry parameters within the refined crystal structure were therefore regarded by Mogul as being not unusual, with enough relevant data found for each parameter to be confident in the results.
The molecular geometry is therefore not seen to deviate from what is chemically reasonable and we can turn our attention to the molecular packing in the crystal. There are essentially two different hydrogen bonds in sulphathiazole phase I, these are an amine to oxygen (sulphonamide) interaction (Figure 4, left) and a thiazole to nitrogen (sulphonamide) contact (Figure 4, right).
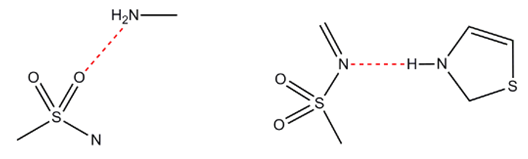
To investigate how reasonable the packing is, we can use the CSD-Materials module of Mercury to determine how often these specific hydrogen-bonded interactions occur in CSD and, more crucially, find out their frequency of occurrence (the number of occurrences as a percentage of the number of structures in which it could occur). The first interaction (amino to sulphonamide oxygen) occurs in 282 crystal structures in the CSD, which works about to be 68% of all the possible observations – this means that the hydrogen bond generally forms when it has the opportunity to do so. An alternative interaction involving the amino group as a donor could be the amino to sulphonamide nitrogen hydrogen-bond – this is also seen to occur in the CSD, but with only a 31% frequency of occurrence.
The second observed hydrogen bond (thiazole to sulphonamide nitrogen) exhibits a frequency of occurrence of 49% compared to the alternative interaction (thiazole to sulphonamide oxygen) which only occurs 21% of the time. We can conclude from these motif searches that the refined structure contains highly likely hydrogen-bonding interactions, which serves to further validate the correctness of this structure. The alternative hydrogen-bonding interactions are also seen to occur relatively frequently in the CSD though, so it is still possible that other stable or metastable packing patterns of the molecule exist, this suggests the potential for polymorphism.
Although the observed hydrogen-bonds correspond to highly likely interactions, based on CSD motif searches, it is important to also look at the actual geometry of each contact. A structure may appear to contain a genuine hydrogen bond, but if the interaction geometry is very poor, this may be an indicator of issues with the refinement. Figure 5 (left) shows an IsoStar plot of close polar X-H contacts (X=N, O or S) with a sulphonamide group in the CSD. Looking at the hydrogen-bonding interactions to one of the sulphonamide groups in the high pressure structure of sulphathiazole-I (figure 5, right), we can see qualitatively that each interaction is situated in a commonly observed region of the IsoStar scatterplot.
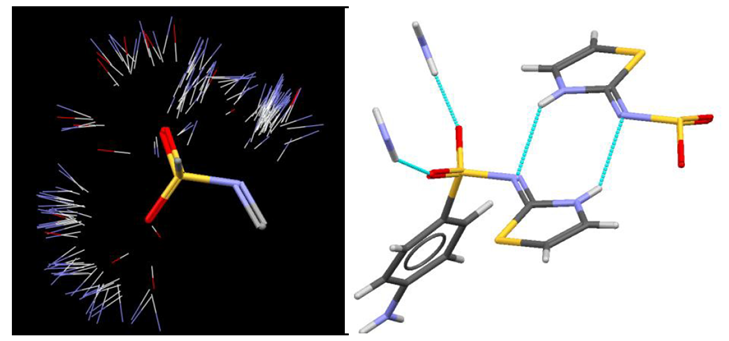
Analysing the geometrical parameters of each of the crystallographically inequivalent hydrogen-bonds we see that the donor to acceptor distances range from 2.36 to 2.98 Å with D-H…A angles between 138 and 172°. The substantial range of geometries for these relatively equi-energetic hydrogen-bonds is an indicator that there may be problems with the data, but each contact is still within the distribution of observed CSD parameters, so the structure is likely to be low-resolution rather than incorrect.
The tools applied here will not indicate definitively whether a structure is correct or not, but they will highlight potential problems or inconsistencies with a structure thus providing an analysis of the relative risk that the structure may be wrong. All the evidence relating to the intramolecular and intermolecular geometries suggests that the structure is indeed reasonable. In this case, the structure was collected at extreme conditions and roughly 60% of the reflections were missing from the collected dataset, but in spite of the poor data quality the refined structure is essentially correct.
Conclusions
On the basis of the intramolecular and intermolecular geometry analyses, it would appear that this high pressure structure is essentially valid even though the underlying diffraction data is of relatively poor quality. This example presented has highlighted the benefits of efficient and thorough structure validation tools in making a decision about the reliability of an individual crystal structure.
References
1. G. J. Kruger, G. Gafner, Acta Crystallogr., Sect. B 1972, 28, 272-283.
2. T. N. Drebushchak, E. V. Boldyreva, M. A. Mikhailenko, J. Struct. Chem., 2008, 49, 84-94.
3. A. Dawson, D. R. Allan, S. Parsons, M. Ruf, J. Appl. Crystallogr., 2004, 37, 410-416.
5. F. H. Allen, Acta Crystallogr., Sect. B, 2002, 58, 380-388.
Next Steps
Learn more about the Cambridge Structural Database.
Learn more about Mogul, Mercury and IsoStar.
Follow the link to our self-guided workshops demonstrating the tools included in the CSD-Materials suite. You’ll get the chance to try out the example problems!
To discuss further and/or request a demo with one of our scientists, please contact us via this form or .