Undruggable targets—a data-driven strategy to reveal binding sites
It is estimated that 85% of the human proteome is considered undruggable(1)—finding effective ligands (pharmaceuticals) to target these proteins is considered exceptionally hard, or impossible.
Here we pose that this may not be impossible! We examine what makes a protein “undruggable”, and present strategies which can be used against such targets.
What makes a protein undruggable?
Traditional drug design focuses on the isolation of a specific protein, or target. The binding site of the target is identified, and various strategies can be used to design molecules that will bind to the pocket, giving a therapeutic response. Undruggable proteins cannot be targeted by this approach. This can be due to a variety of factors, some of which are explored below.
- Total inhibition: binding site occupation results in a complete loss of function, including healthy function levels of the protein.(2)
- Competition: orthosteric inhibitors may require very high concentrations to outcompete natural ligands, resulting in off-target effects and potential toxicity or tolerance issues.(2)
- Pocket: lack of structured or well-defined pocket to accomodate a drug—genetic downregulation is required.(2)
- No hits in screening: if no leads are identified when screening against commercial molecular libraries, this can make it appear the protein is undruggable.(3) Potential solutions to this issue are discussed below.
- Protein-protein interactions: where preventing a protein-protein interaction is the target, there are often very large interaction surfaces with no obvious pockets.(4) Again we explore potential solutions below.
What strategies are used to target undruggable proteins?
So, are some proteins really undruggable? Or is it simply that the right technology or approach has not yet been found. These techniques are already in use and producing results against proteins that were previously considered undruggable.
Modulation
Instead of inhibiting a protein, selective modulation can be used. This may include depletion (protein degradation), rescue (for example the design of chaperone molecules that manage protein folding), or amplification.(2) This approach does not specifically target the undruggable protein, but instead asks; which other proteins in the protein–protein interaction network could be targeted to produce the desired effect?
Expansion of chemical space
This approach is effective against two of the problems that make some proteins “undruggable”; no hits in screening, and protein-protein interactions.
If leads are not identified from commercial libraries, this can make it appear that the protein is undruggable. But this could be a limitation of the library, as discussed by Dandapani and Marcaurelle.(3) Increasing the number and variety of molecules available in libraries is important, but a lengthy process. Fragment screening is an alternative approach.
Fragment screening
By screening against fragments, rather than larger molecules, more opportunities are made to identify interactions. In our recent interview with Dr Mihaela Smilova, she explained that chemical space is estimated to increase eight-fold for every heavy atom added to a molecule. By screening fragments higher hit rates and more opportunities are found when compared to commercial library screening.
Fragment screening has also been demonstrated as effective in targeting protein–protein interactions. For example, BCL-2 whose interactions with other proteins are involved in cancer pathways. Fragment-based drug discovery was employed when traditional library screening failed, and, with further optimization, yielded a molecule with nanomolar activity against the target.(5)
Tools such as hotspot mapping automate this process in silico. This script highlights visually on a protein where fragments are likely to bind, based on big-data learnings from experimentally derived data in the Cambridge Structural Database (CSD) and Protein Data Bank (PDB).
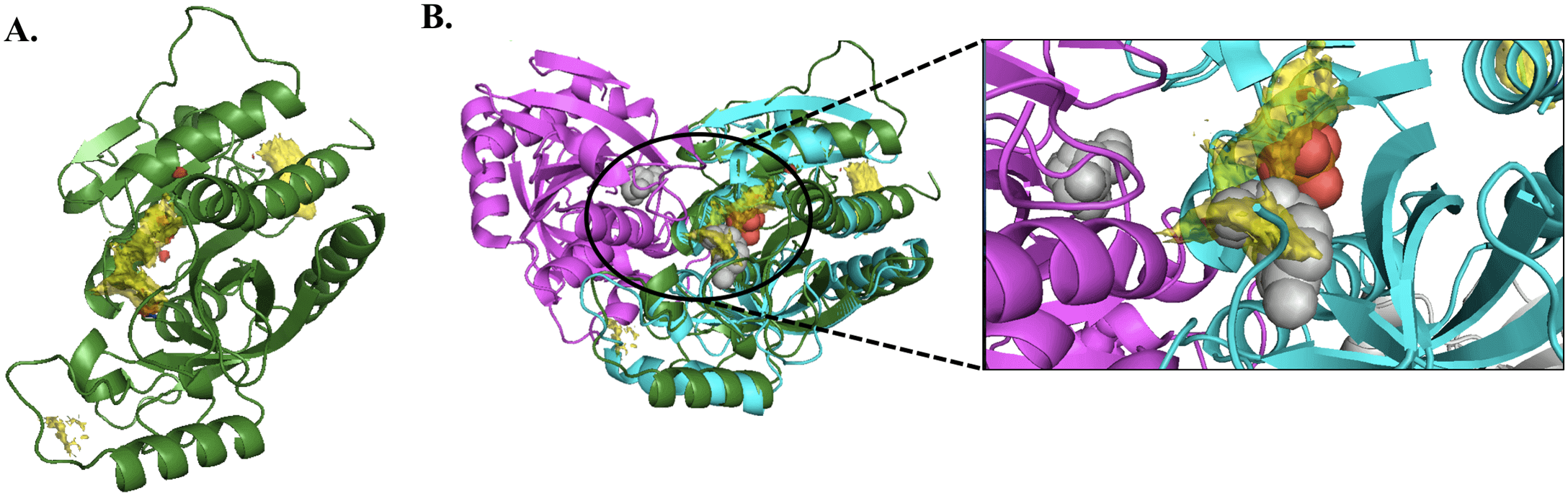
Example of in silico technique hotspot mapping, which models fragment binding to identify druggable sites on a target protein. Fragment binding is commonly used against “undruggable” targets. Image shown from Malhotra et al., https://doi.org/10.1371/journal.pntd.0005883.g006
This data-driven approach has been successfully employed by scale-up drug discovery company ExScientia in their workflows to design novel therapeutics. It has also found success across the literature, for example to assess the druggability of allosteric sites of AChE, an enzyme identified in Alzheimer’s disease pathways,(6) or to predict druggable sites of ML2177c, a potential target in bacterial infections including leprosy.(7)
Learn more about hotspot mapping in the whitepaper here, including case studies from the literature.
References;
(1) Targeted protein degradation by PROTACs, T. K. Neklesa et al., Pharmacol Ther., 2017, https://doi.org/10.1016/j.pharmthera.2017.02.027
(2) Exploiting Folding and Degradation Machineries To Target Undruggable Proteins: What Can a Computational Approach Tell Us?, S. A. Serapian et al., ChemMedChem., 2021, https://doi.org/10.1002/cmdc.202000960
(3) Grand Challenge Commentary: Accessing new chemical space for ‘undruggable’ targets, S. Dandapani et al., Nature Chemical Biology, 2010, https://doi.org/10.1038/nchembio.479
(4) Emerging Trends in Cancer Drug Discovery—From Drugging the “Undruggable” to Overcoming Resistance, J. Rudolph et al., Cancer Discov, 2021, https://doi.org/10.1158/2159-8290.CD-21-0260
(5) The Challenge of Drugging Undruggable Targets in Cancer: Lessons Learned from Targeting BCL-2 Family Members, G. L. Verdine et al., Clin. Cancer. Res., 2007, https://doi.org/10.1158/1078-0432.CCR-07-2184
(6) Identification of new allosteric sites and modulators of AChE through computational and experimental tools, C. Roca et al., J Enzyme Inhib Med Chem., 2018, https://doi.org/10.1080/14756366.2018.1476502
(7) Decoding the similarities and differences among mycobacterial species, S. Malhotra et al., PLoS Negl. Trop. Dis., 2017, https://doi.org/10.1371/journal.pntd.0005883