Mogul in Action: Astex Pharmaceuticals. Protein–Ligand Complex Analysis to Identify Unusual Torsional Geometry in Drug Candidates
Here we highlight a paper by John W. Liebeschuetz from Astex Pharmaceuticals where Mogul was used to analyze the geometry of drug-like ligand models for high-resolution crystallographic protein-ligand complexes. This is part of our series highlighting examples of the Cambridge Crystallographic Data Centre (CCDC) tools in action by scientists around the world.
Summary
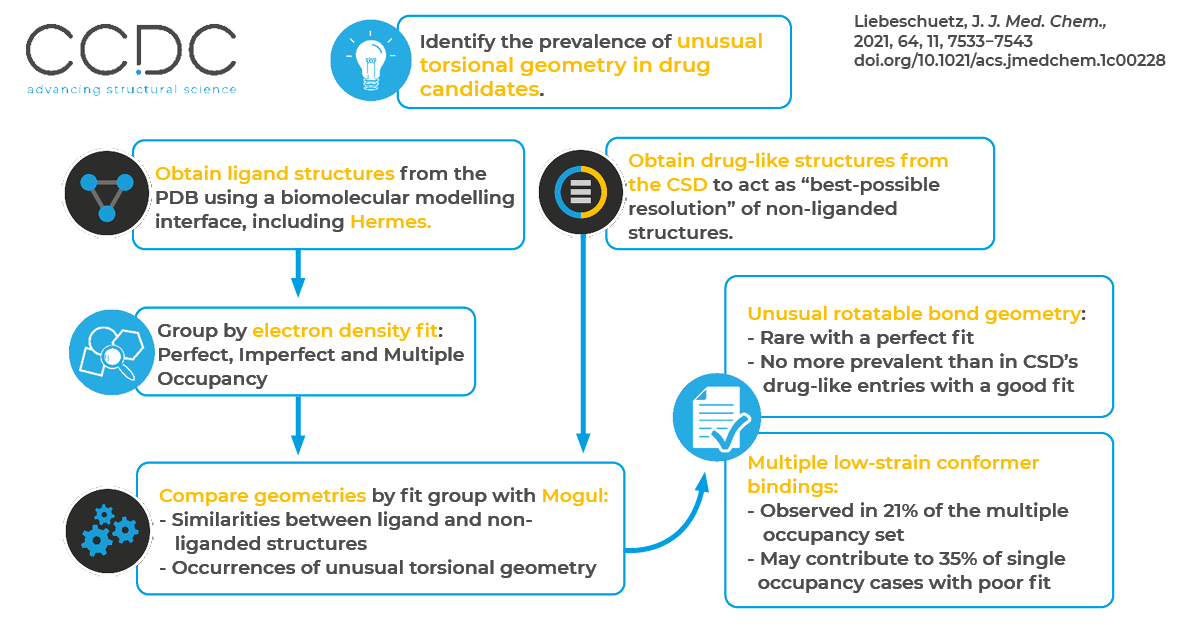
In this work, Liebeschuetz used Mogul to investigate the prevalence of unusual torsional geometry of drug-like ligand models for high-resolution (<1.1 Å) crystallographic protein-ligand complexes. Mogul is a knowledge-based library of molecular geometry derived from the Cambridge Structural Database (CSD).
Based on this analysis, unusual rotatable bond geometry is rare in crystallographic ligand models with well-fitted electron density maps and is no more prevalent than in the CSD’s unliganded, drug-like entries. When unusual torsional geometry does occur with a complex solved with high-resolution and a perfect electron density fit, it’s usually associated with strong polar, metal or covalent ligand-protein interactions.
Unusual torsional geometry is more prevalent when the fit to electron density is imperfect. One reason: a refined ligand structure is only one out of a collection of structures that can each fit the electron density adequately well with an imperfect fit. In this analysis, multiple low-strain conformer bindings were observed in 21% of multiple occupancy structures thought to be solved with a structure displaying more unusual geometry. This may also lie behind 35% of single-occupancy cases that have a poor fit to the electron-density map. Liebeschuetz concluded that multiple conformer-ligand binding constitutes an under-recognized phenomenon in structure-based drug design, and more robust crystallographic refinement methods are needed to handle such cases.
Why
Usually, successful drugs are efficient ligands for their sites of action and introduce little conformational strain when they bind since any energy required to achieve the bioactive conformation adversely affects the binding affinity at the active site. Realistically, however, perfectly optimizing both the ligand geometry and the fit to the expected binding site isn’t possible. Some conformational strain is expected, with low conformational strain being the goal. To achieve low conformational strain, the geometric features – the bond lengths and angles, heavy atom dihedral angles of rotatable bonds and ring geometries – within bound ligands should not differ greatly from the same geometric features in the unbound molecules. But how much strain is acceptable?
By analyzing the geometries of perfect, high-resolution structures with Mogul, Liebeschuetz was able to find that a torsional strain greater than 2 kcal mol−1 is unlikely. The higher prevalence of torsional strain in both high- and low-resolution structures with imperfect electron-density mappings is potentially due to some artefact or multiple conformer-ligand binding.
How
Mogul provides precise information on preferred molecular geometries by accessing millions of chemically classified bond lengths, valence angles, acyclic torsion angles and ring conformations derived from the CSD. This tool enabled Liebeschuetz to compare the reported prevalence of unusual geometries in three different sets of crystallographic data.
The first set contained 83 complexes from high-resolution (<1.1 Å) structures from the Protein Data Bank (PDB) that were manually grouped further into three categories: multiple occupancy, perfect and imperfect. To be classified as “perfect,” no substructure could lack close-fitting electron density, and no significant unassigned electron density existed adjacent to the ligand. For the high-resolution set, this meant every heavy atom had to have discrete regions of electron density centred on it.
The second dataset contained 79 structures based on moderate resolutions (2.4−2.8 Å) and was further subdivided like the high-resolution set.
The third set comprised 66 drug-like structures from the CSD and served as a reference set of “best-possible resolution” of non-liganded, drug-like structures. He focused on the high-resolution structure set.
Read More
Read the full paper: “The Good, the Bad, and the Twisted Revisited: An Analysis of Ligand Geometry in Highly Resolved Protein-Ligand X‑ray Structures,” J. Med. Chem., 2021, 64, 11, 7533−7543.
Learn more about how Mogul enables you to rapidly validate a structure’s complete geometry and identify any unusual features.
Explore more examples of CCDC tools in action.
To discuss further and/or request a demo of Mogul with one of our scientists, please contact us via this form or .