Optimize Crystal Structures with Experimental PXRD Data
Crystal structures can now be optimized using an experimental diffractogram using AutoFIDEL functionality. This is available via the Mercury powder menu, for CSD-Materials and paid academic customers.
AutoFIDEL, previously only available in the CSD Python API, is particularly useful for both predicted crystal structures and low-temperature experimental crystal structures, where the powder pattern is measured under significantly different conditions, which can result in differences in lattice parameters and powder patterns. It builds on the matching functionality provided in Mercury v.2025.1 to deliver a powerful yet intuitive tool for PXRD analysis and structure solution.
What is AutoFIDEL?
AutoFIDEL is Jonas Nyman’s implementation of the FIDEL method using similarity measures developed by de Gelder et al.. It streamlines the process of comparing powder diffraction patterns to potential structures and then optimizes matches for better accuracy.
How Can I Use It?
Importing an experimental file (in .xy, .xye or .xrdml formats) into the Powder Pattern dialog enables initial matches to be evaluated. Users can quickly click through structures of interest loaded in the Structure Navigator to identify patterns that show a good starting match using both visual checks and the Similarity figure provided, a value between 0 and 1 where 1 is a perfect match. However, this match is unlikely to be perfect, particularly where the structure of interest was measured or predicted at a different temperature.
The Powder Pattern dialog now presents users with the ability to optimise the loaded structure in the visualizer against that imported pattern using the Optimise button. This will optimise the parameters selected in the Customise dialog, which can include lattice parameters, molecular position, intramolecular geometry and preferred orientation. The algorithm will then seek the best possible match with the experimental crystal structure using the Similarity figure.
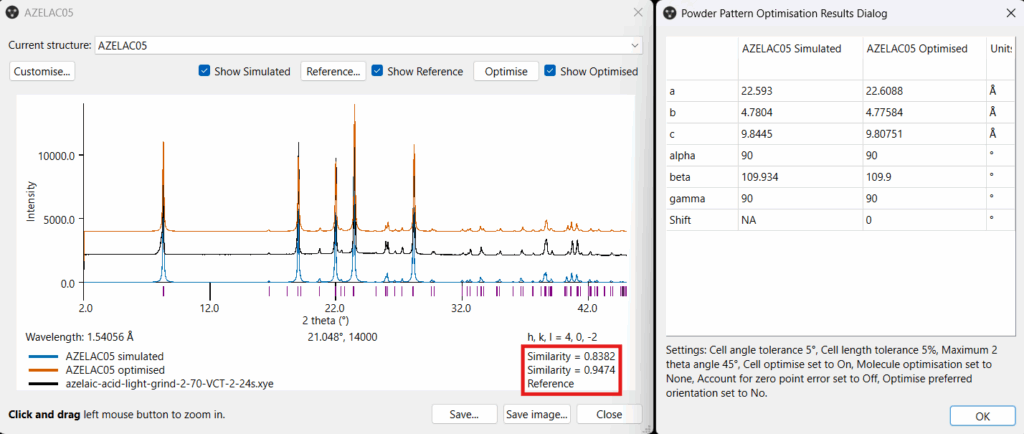
The method can be used with routinely collected PXRD data in combination with Crystal Structure Prediction data to rapidly identify crystal structures.
Acknowledgements
We are grateful for the papers of Habermehl et al. and de Gelder et al. for the principles used in AutoFIDEL. We have done extensive validation on the methodology, thanks to the contributions of experimental patterns by a number of our collaborators, including Anuradha Pallipurath from the University of Leeds, Martin Ward from CMAC/University of Strathclyde and David Coates from Eli Lilly. Erin Johnson from the University of Dalhousie/Cambridge, R. Alex Mayo from the University of Dalhousie/Ottawa and Alberto Otero de la Roza from the University of Oviedo, have supported development through useful discussions on theory and implementation.
Next Steps
Contact us here to discuss further and/or request a demo with one of our scientists.