Enhanced Powder Pattern Matching, Optimization, and Simulation
In CSD-Materials, new functionality in the CSD Python API, based on AutoFIDEL algorithms, matches and optimizes crystal structures based on experimental powder diffraction patterns, tolerating noisy, shifted patterns, or those with preferred orientations.
Accurately matching and optimizing crystal structures is a critical task. The process involves comparing experimental patterns with predicted ones, which can help identify the most likely structure of a material. This method becomes particularly important when dealing with polymorphs or complex landscapes of potential structures.
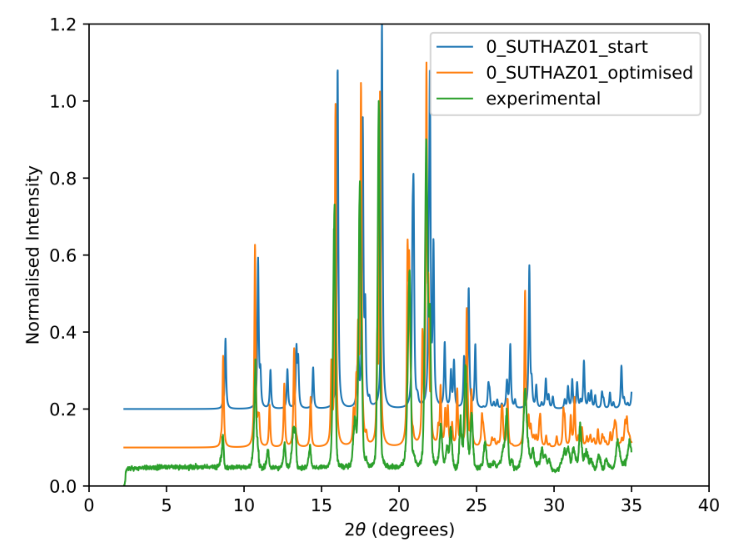
Sulfathiazole optimized using experimental data
What is AutoFIDEL?
AutoFIDEL is Jonas Nyman’s implementation of the FIDEL method using similarity measures developed by de Gelder et al.. It streamlines the process of comparing powder diffraction patterns to potential structures and then optimizing matches for better accuracy.
Powder Matching: The First Step
The first step involves the matching of experimental powder patterns with simulated ones. Typically, you begin with an experimental pattern, which might come from a sample you’ve analysed or even from a published journal article. The goal is to see if this pattern can be matched with any known structures.
You may have a list of possible structures, which could include different polymorphs or a variety of configurations generated through computational crystal structure prediction methods (CSP). You can now simulate the powder X-ray diffraction (PXRD) patterns for these structures and compare them to your experimental data. This process helps identify which structure, if any, aligns most closely with the experimental pattern.
Traditionally, many users rely on manual overlay techniques. However, AutoFIDEL automates this process, utilizing sophisticated algorithms to determine the best match more objectively and efficiently.
The Optimization Process: Refining the Match
Once a structure has been identified that closely matches the experimental pattern, the next step is optimization. The initial match might not be perfect, and slight adjustments to the structure could lead to a much better fit. This is where AutoFIDEL’s optimization capabilities come into play.
The optimization step involves fine-tuning various parameters of the structure to maximize its similarity to the experimental pattern. This is done by adjusting lattice parameters and preferred orientation settings. The goal is to achieve the highest possible match, quantified by the PXRD similarity measure.
This optimization process not only saves time by automating what would otherwise be a manual, trial-and-error process but also improves the accuracy of the structural analysis, providing a more reliable identification of the material’s structure. This method allows the user to easily solve structures from routine lab PXRD and crystal structure prediction.
How to Access the New Functionality
The initial implementation is in the Python API. You can find and run a sample script and in the GUI in Mercury under the ‘CSD Python API > Prototypes’ menu.
A CSD-Materials license is required to access the AutoFIDEL functionality.
Acknowledgements
We have done extensive validation on the methodology, thanks to the contributions of experimental patterns by a number of our collaborators, including Anuradha Pallipurath from University of Leeds, Martin Ward from CMAC/University of Strathclyde and David Coates from Eli Lilly. Erin Johnson from University of Dalhousie/Cambridge and R. Alex Mayo from University of Dalhousie/Ottawa and Alberto Otero de la Roza from University of Oviedo, have supported development through useful discussions on theory and implementation.
Improved PXRD Simulation
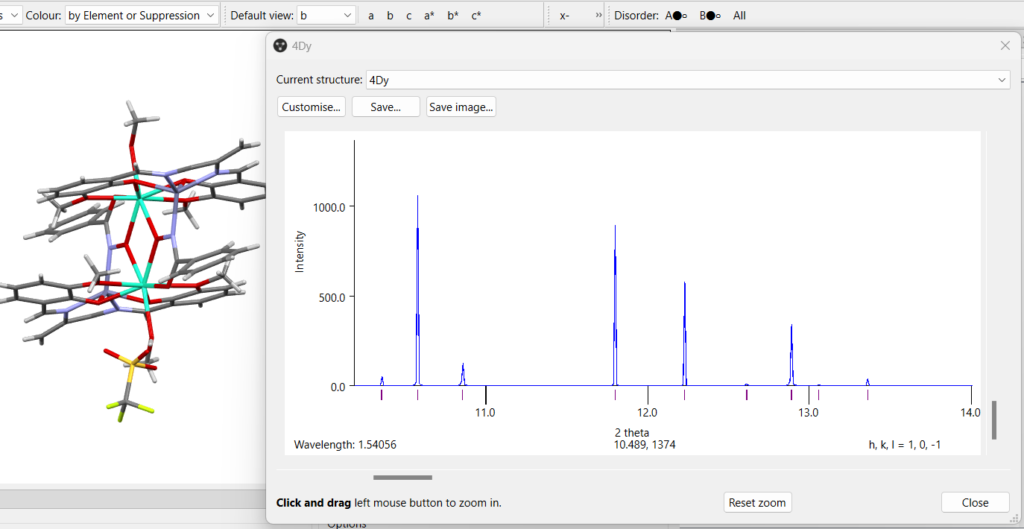
For all users including CSD-Community users, the simulated powder X-ray diffraction patterns in Mercury have also been improved, including:
- Handling of different disorder assemblies
- New variable slit type
- Smoother curves and scroll bar on zoom
- *.xy and *.xrdml file formats are now supported when exporting simulated patterns or when reading in patterns via the CSD Python API
- Settings are persistent from one session to the next
- Handling of partial occupancies for symmetry generated atoms
- For customers who have access to preferred orientation, predicted orientation information based on BFDH morphology
- Ease of overlay – the images have, where possible, a transparent background.
Next Steps
- Watch On-Demand Webinar: How To Match Powder Patterns to Crystal Structures
- Ask a question or request a demo with one of our scientists here, or
- More information on the CSD Python API and Mercury.
- More information on CSD software trusted by academic and industrial institutions around the world.
- Check out the Cambridge Structural Database for yourself and see how your research can benefit from the combined knowledge of over 1.3M small-molecule organic and metal-organic crystal structure data.
- See case studies of the CSD in action, driving forward the boundaries of scientific research.