Investigating Solid Form Stability: Understanding Hydrogen Bond Propensity in Mercury
This blog covers content from a CCDC Virtual Workshop and focuses on Hydrogen Bond Propensity, a component within CSD-Materials that studies the hydrogen bond network in a crystal structure. The full workshop can be accessed here.
Exploring Hydrogen Bonds Through Mercury
Mercury allows users to view, analyse, and understand molecular structures and their properties. By exploring 3D structures, the user can get a deeper understanding of the geometry of molecules, the way molecules pack together, and what voids and channels exist within a structure. Important information about the characteristics that influence stability can be gained, such as molecular conformation, hydrogen bond geometry, hydrogen bond donor/acceptor pairing, and other intermolecular interactions present in the structure.
To view hydrogen bonds in a structure in Mercury, the user should tick “H-Bond” and “Show Hydrogens” in the Display section, Figure 1. The default H-bond definition can be changed, and the hydrogen bonds can be coloured by distance (Display > Colours > Contacts > Colour by distance).
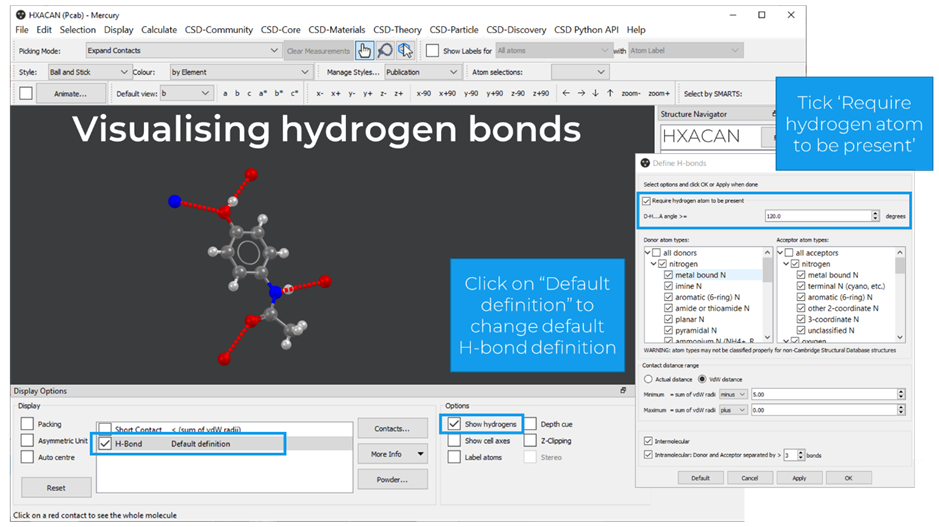
The contacts can be expanded by selecting “Expand Contacts” as the picking mode on the top left of the visualizing area, and then by clicking on atoms at the end of the dashed lines.
To learn more about the basics of Mercury, follow the link to our CSDU on-demand, free course “Visualization 101 – Visualizing structural chemistry data with Mercury”. To find out more on how to define and visualize hydrogen bonds in Mercury, watch the video tutorial.
Can Structural Knowledge Mitigate Risk?
An example of the impact of different interaction networks on drug solid forms dates back to the 1990s, and involved the antiretroviral HIV drug ritonavir. After being released in the market, ritonavir had to be quickly withdrawn because its solid form changed, having a devastating impact for patients that were relying on the drug for treatment. Some CCDC components, like the Hydrogen Bond Propensity, can now be used to mitigate the risks of that happening again by helping to improve the understanding of the stability of a solid form before it goes to market. By using the wealth of information in the CSD, predictive analytics can identify the likelihood of specific molecular interactions occurring from similar crystal structures.
Mercury and Hydrogen Bond Propensities Overview
Hydrogen Bond Propensity (HBP) is a component that is available in Mercury. It identifies all the possible hydrogen bonds and their networks, and can assess the contribution of each Donor-Acceptor (D-A) pair to the observed network. HBP can also rank donors and acceptors in the molecule, generate a landscape of putative H-bond networks, and compare hydrogen bonding in polymorphic structures. This component can be used with neutral molecules, salts, and co-crystals. Polymorphic families with Z’ = 1 (number of molecules in the asymmetric unit equal to 1) can also be plotted in the same HBP landscape.
To use the HBP component, the user should open the structure in Mercury and go to the Propensity Prediction wizard. This is accessible through the path CSD-Materials > Polymorph Assessment > Hydrogen Bond Propensities. A 2D diagram of the molecule that was selected in Mercury will then appear, alongside a list of hydrogen-bond donors and acceptors included in that molecule, and a list of functional groups from the library available in Mercury for HBP (Figure 2). The component matches each of the donors and acceptors to the functional groups present in the library (Figure 2).
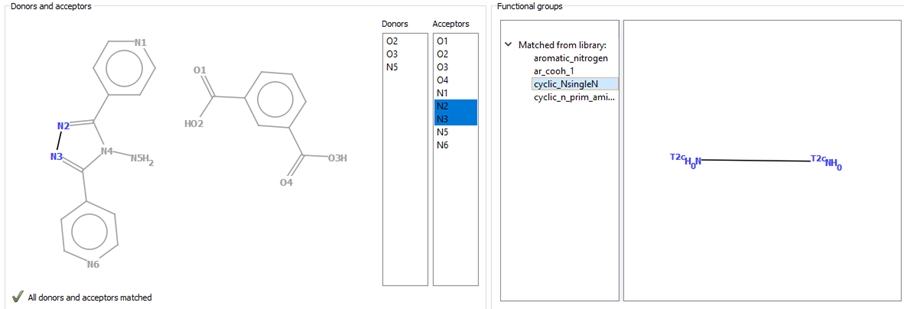
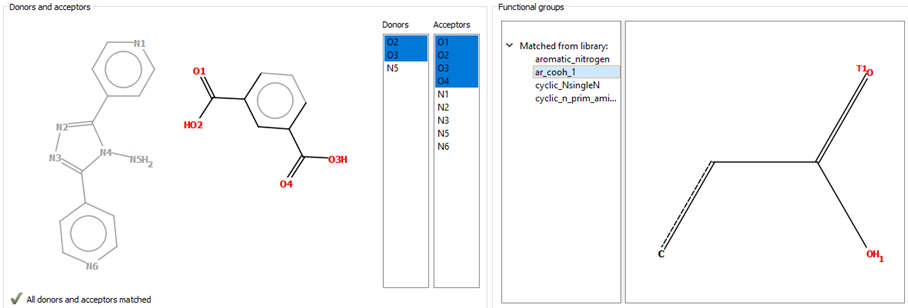
After the functional groups are matched, the CSD will be searched to find the molecules that contain these functional groups (ideally more than one per molecule). This can be done from the “Generate Fitting Data” window by pressing “Generate” (Figure 3). We recommend allowing the program to fully finish each search.
The ideal search would lead to an even coverage of functional groups, with a minimum count of 300-500 observations per functional group. Once the search is performed, the wizard will do a smart selection, picking a reasonable number of hits for each functional group. The slider can be used to increase or decrease the number of hits (Figure 3). At this step, the program saves the list of matching entries as a gcd file, a plain text list of CSD Refcodes that can be loaded into Mercury.
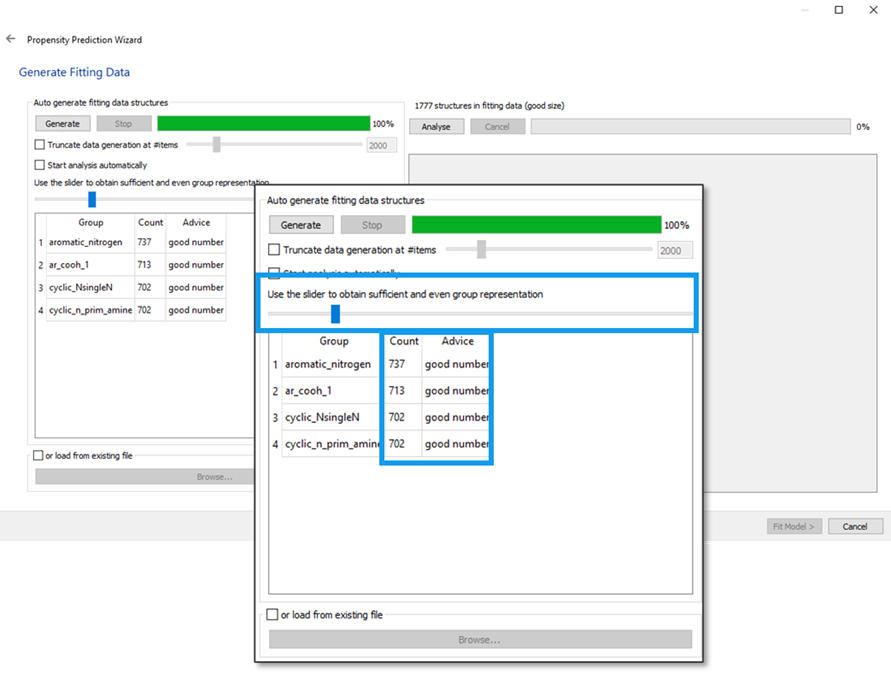
The next step consists of analysing the data extracted from the CSD, and this can be done by clicking “Analyse” from the “Generate Fitting Data” window. If any group has very low numbers highlighted in red on either True or False, the user should make sure the “Ignore?” checkbox is ticked.
A logistic regression is then performed on the fitting data to obtain all the propensity values by clicking on “Fit Model”, and the model can be refined by omitting variables with no significance.
The final window of the HBP search is reported in Figure 4 and is obtained by clicking “Accept & Calculate”. This window includes three components: the landscape on the left, the propensity scores in the middle, and the co-ordination scores on the right (Figure 4).
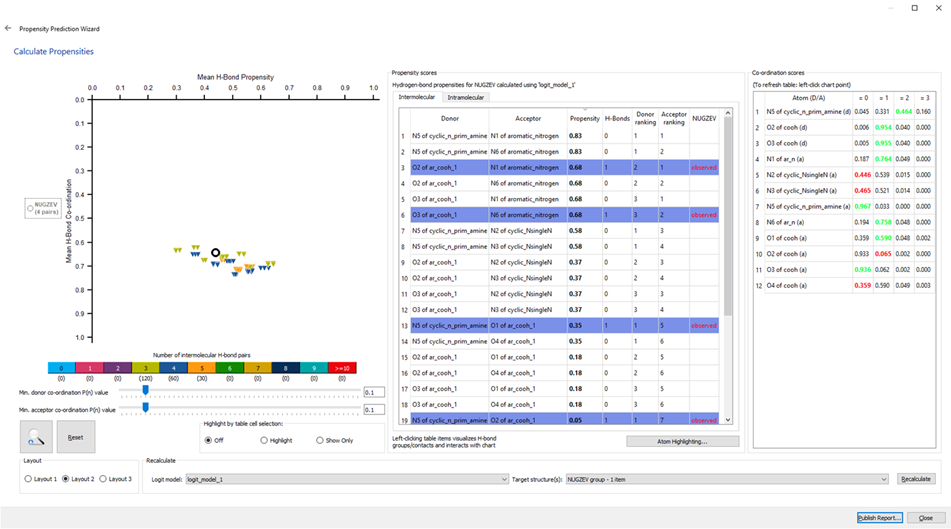
In the propensity scores window, there are two tabs: one for intermolecular interactions and one for intramolecular interactions. Significative information regarding the propensity scores of each possible combination of donors and acceptors can be gained in this step, representing the likelihood that a specific hydrogen-bond forms between the two selected functional groups.
The coordination scores window contains information on how each donor or acceptor behaves compared to what is more commonly present in the CSD. For example, the first entry in Figure 4 is a primary amine that has two donor protons and it can be seen that, as expected, it forms two hydrogen-bonds.
The coordination scores in the chart are colour coded to indicate how the donor and acceptor atoms’ behaviour in the active network compares to observations in the CSD. The green highlighted values indicate that the atom is following the expected trend, while the red values represent deviations from the expected behaviour. For example, row 10 in the list in Figure 4 identifies oxygen atom O2 of a carboxylic acid which strongly prefers to not participate in any hydrogen bond interactions, but that is involved in one bond in the active structure.
In the HBP landscape chart, the putative structures can be visualized along with the observed structure, represented by the white circle. The networks are colour coded according to how many hydrogen bonds are present in them. The higher Mean H-Bond Propensity values indicate the network involves hydrogen bond pairs that have a high likelihood of happening, and higher Mean H-Bond coordination values show that the pairings in the network follow the trends observed in the CSD.
The HBP analysis can be saved as a HTML report from the Propensity Prediction wizard by clicking “Publish Report”. Additionally, the landscape is saved as an image (REFCODE.png) and can be re-plotted with the data in REFCODE_chart_data.tsv, while the HBP analysis can be reopened in Mercury loading the regression model (REFCODE_regression_results.txt) in case the user wants to perform further analysis without re-running it from the start.
Full Interaction Maps Overview
Full Interaction Maps (FIMs) are maps of preferred sites for interactions around a molecule calculated using CSD data and can complement the HBP analysis.
As can be seen in Figure 5, this component is accessible via Mercury, through the path CSD-Materials > Full Interaction Maps. Different probes can be used to study the most common intermolecular interactions (Figure 5): for example, probes for hydrogen-bond donors, acceptors and hydrophobic interactions are available.
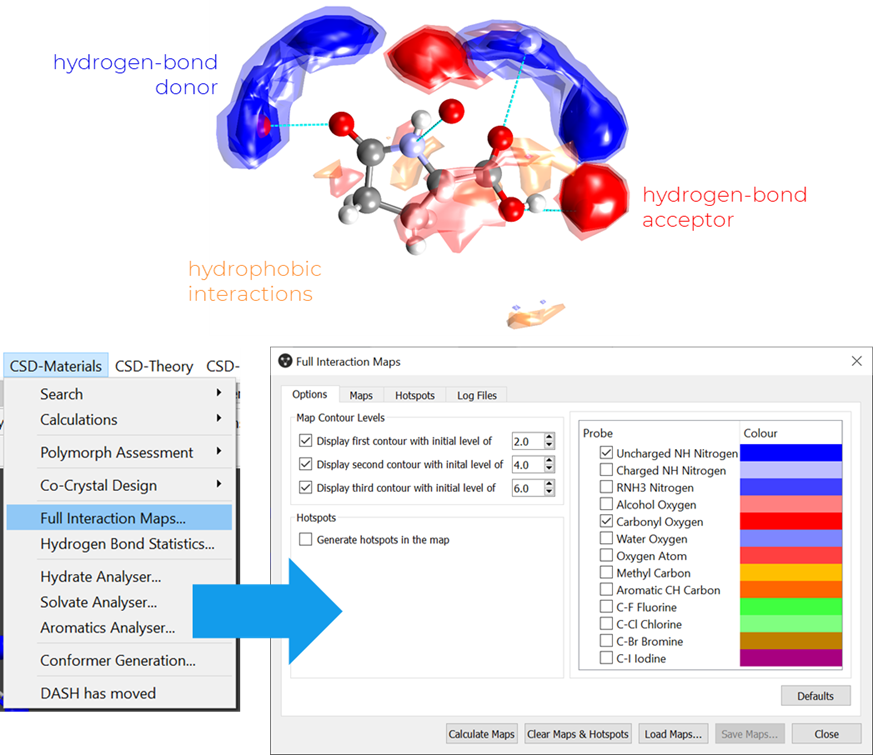
One important aspect of FIM analysis is that, for multi-component systems and crystal structures with Z’ > 1, the map needs to be calculated separately for each of the chemical components.
To learn more about the basics of FIMs, see our CSDU on-demand, free course “Analysing intermolecular interactions 101 – Full Interaction Maps”.
Featured Questions and Answers Asked at the Workshop
If we don’t have the molecules in the CSD how do we calculate the HBP?
You can load the structure that you want to analyse into Mercury, if you have your structure as a .cif file or a .mol2 file, for example. You can also sketch your molecule (under File -> Sketch Molecule, in Mercury). The CSD is then used as a source of knowledge about hydrogen bond interactions, but your structure does not have to be in the database in order to be analysed.
How can I generate the landscape including more than one polymorph?
You can load in multiple structures on the final wizard window – at bottom right there is a place to select your Target Structures. From that dropdown menu you can select more than one structure. However, it only works for systems with one molecule in the asymmetric unit and where the Z’ is the same for the structures, so it doesn’t work for multicomponent structures. The example chart shown above was generated using the Python API.
Can you use HBP analysis to rationalize the difference between a salt and a cocrystal of a material which uses the same coformer species?
Technically it is possible. You can input your charge species and the data that is collected will involve charged groups and output will result for that. You can do it then for neutral components, so the information is sourced now for neutral structures, so the results will be different.
Next Steps
To discuss further and/or request a demo with one of our scientists, please contact us via this form or .