How to Search, Visualize, and Analyse MOF Structures
With the increasing demand for efficient porous functional materials for carbon capture, moisture harvesting, and pharmaceutical uses, the global market of MOFs is forecasted to grow significantly in the upcoming years.
Despite this, several factors are still impacting a wider use of MOFs, preventing them from reaching their full potential. To expand this topic, read our blog “What Is Stopping MOFs Being More Widely Used?”.
In this blog we present some of the functionalities included in CSD-Frameworks that help optimize the design and development of new functional porous materials, like MOFs.
We will start by exploring how to search for MOF structures in the Cambridge Structural Database (CSD) and then learn how to visualize and analyse porous crystal structures from proprietary or published data.
Read our white paper Accelerating Porous Materials Design and Development—A Data-Driven Approach to learn more.
Searching for MOF Structures in the CSD
The development of new functional materials often starts with the study of the landscape of existing compounds. With over 128,000 experimental MOF-like structures, the CSD represents a trusted source that provides big-data insights into the structural and physical properties of existing MOF materials.
To search for MOF structures in the CSD:
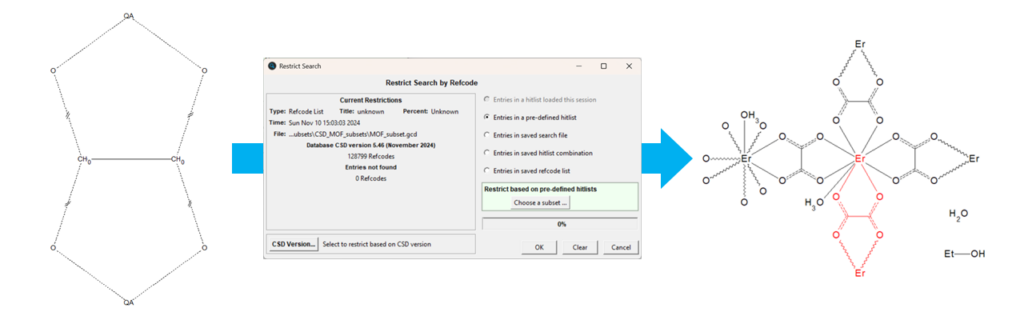
- Open the CCDC’s software ConQuest and click “Draw”;
- Draw a sketch of the chemical connectivity that you want to search in the database, such as the one reported in Figure 1 for an oxalate ligand bridging two metal atoms;
- You might want to expand your search by including more than just one metal centre: to do this, right click on the metal, then click “Element” > “More” > “Other Elements”; by choosing “Multi Pick” as picking mode, you can select multiple periods and groups from the periodic table, or just multiple specific elements of your interest;
- The bond type can also be modified: to do this, right click on the bond, then click “Type” and select a specific type of bond; by selecting “Any”, all the bond types listed will be included, while “Variable Bond Type” allows you to include or exclude any of the bond types listed;
- To limit your search to MOF-like frameworks, click “Search” > “Select Subset” > “Entries in a pre-defined hitlist” > “Choose a subset” > “CSD MOF subsets” > “MOF subset” > “OK” > “Start Search”.
The parameters reported above can be changed according to the specific features you are looking for. For example, to further refine your search to only 3D MOFs, you can choose the “3D MOF subset” instead of the whole MOF subset.
Additionally, the chemical connectivity (under “Draw”) is only one of the searching parameters available in ConQuest. Other searching criteria can be found on the left-hand side of the “Build Queries” menu, and include the unit cell and space group, formula and chemical composition, and the presence of guest molecules.
Finally, separate queries can be combined to make your search even more specific.
Visualizing MOF Structures
Performing an in-depth structural analysis of MOF structures helps understanding and rationalizing structure-property relationships for these materials.
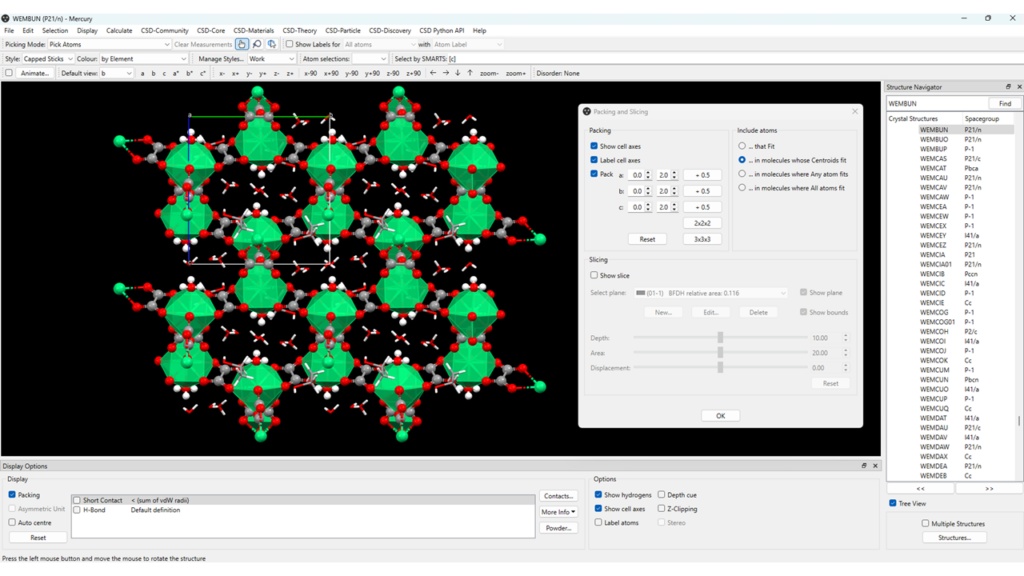
One of the features that you might be interested in visualizing in a MOF structure is its 3D packing. For this example, we will be using the crystal structure of CSD Entry WEMBUN. To analyse its packing:
- Open the CCDC’s software Mercury and search for WEMBUN in the “Structure Navigator” panel;
- A feature that can make it easier for you to visualize the packing later is to select at this stage the style “Polyhedral” for the main framework, then select the solvent molecules and set “Capped Sticks” as style for these;
- Go to “Calculate” > “Packing/Slicing”; make sure that “Pack” is ticked in the “Packing” panel and click on “2x2x2”;
- Now that the structure is expanded, you might want to view it along a specific crystallographic axis, such as the a axis for the case shown in Figure 2.
Other options are available in the “Packing and Slicing” panel to further customize how the 3D packing is visualized.
You can also choose to hide the cell axes and the hydrogen atoms in the structure. Additionally, it might be helpful to show the labels for the metal atoms only, to easily identify the position and type of metal centre.
By clicking “Animate” on the top-left corner of the main Mercury window you can also choose to see the structure rotating or oscillating around an axis.
Finally, images and videos can be extracted and used in scientific articles, a thesis, and in presentations to help you highlight structural features of your MOF structure.
Importing Your Own CIF
While each of the structures in the CSD are edited and enhanced by editors at the CCDC to ensure they have the correct chemical connectivity, if you open your own CIF with Mercury, the bond information will be missing. In this case:
- Go to “Edit” > “Auto Edit Structures” and use the “Identify polymeric bonds” functionality.
Analysing MOF Structures
Voids Analysis
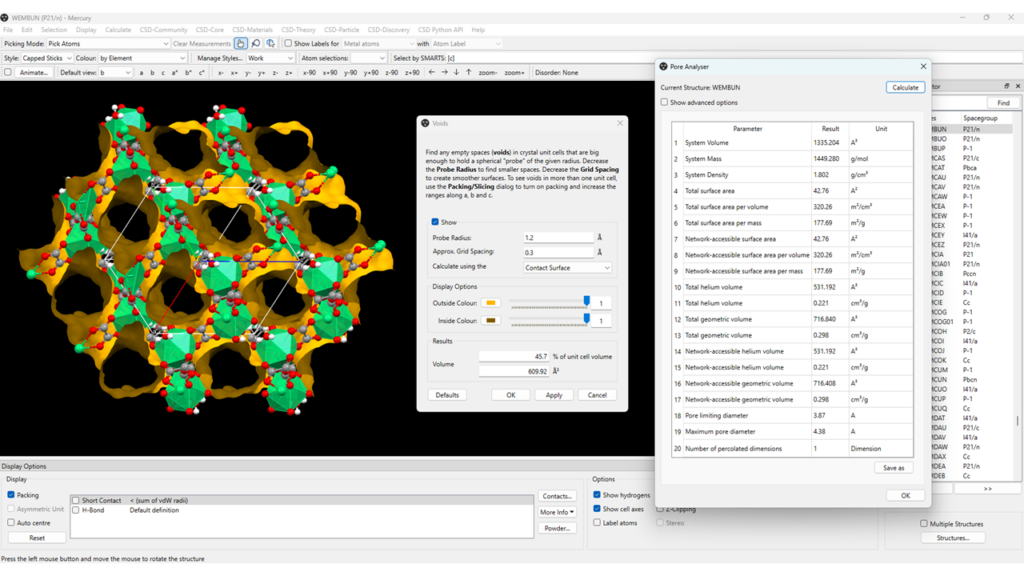
By analysing the packing of WEMBUN reported in Figure 2 it can be seen that channels are present in the structure. To further expand this analysis, Mercury enables the calculation and visualization of voids and pores in the structure. To do this:
- Select the solvent molecules in the structure, go to “Edit” > “Edit Structure” and under “Remove” click on “Selected Atoms”;
- Now that the solvent molecules are removed, go to “Display” > “Voids” > “Apply”;
- From here you can choose to show the packing of the MOF alongside the channels, show the unit cell axes, and rotate the structure to understand the direction and dimensionality of the channels (Figure 3, left);
- The probe radius and grid spacing used for the void calculation, as well as the display options, can be customized from the “Voids” panel (Figure 3, centre);
This analysis calculates the volume of the pores in the structure and allows you to visualize them. However, the Pore Analyser component of Mercury enables the calculation and investigation of the properties of these pores, such as the total pore volume and surface area, the helium accessible pore volume, the system mass, volume, density, and more.
To run the Pore Analyser:
- Go to “Calculate” > “Pore Analyser” > “Calculate”;
A list of properties of the pores will appear, like the one reported in Figure 3, right.
Hydrates and Solvates Analysis
Looking now at the guest molecules that can fill these voids, we have two functionalities available to analyse them: these are the Hydrate Analyser and the Solvate Analyser.
The Hydrate Analyser and the Solvate Analyser help you analyse the properties of space occupied by guest molecules allowing you to understand the volume occupied by each guest molecule. They can also offer insights on the stability of the framework upon guests’ removal, and on the ability of the material to absorb and release water and other solvents.
To run the Hydrate Analyser:
- Search for WEMBUN in the “Structure Navigator” panel; this will allow you to reload the structure with the solvent molecules;
- To calculate the volume of space occupied by water molecules and to visualize the water spaces in the structure of the framework go to “CSD-Materials” > “Hydrate Analyser” > “Water Space” > “Calculate” (Figure 4);
- From the “Hydrate Analyser” panel you can choose whether to show the water molecules or not, you can modify the probe radius and grid spacing used for the calculation, and you can investigate the water hydrogen-bonding motifs present.
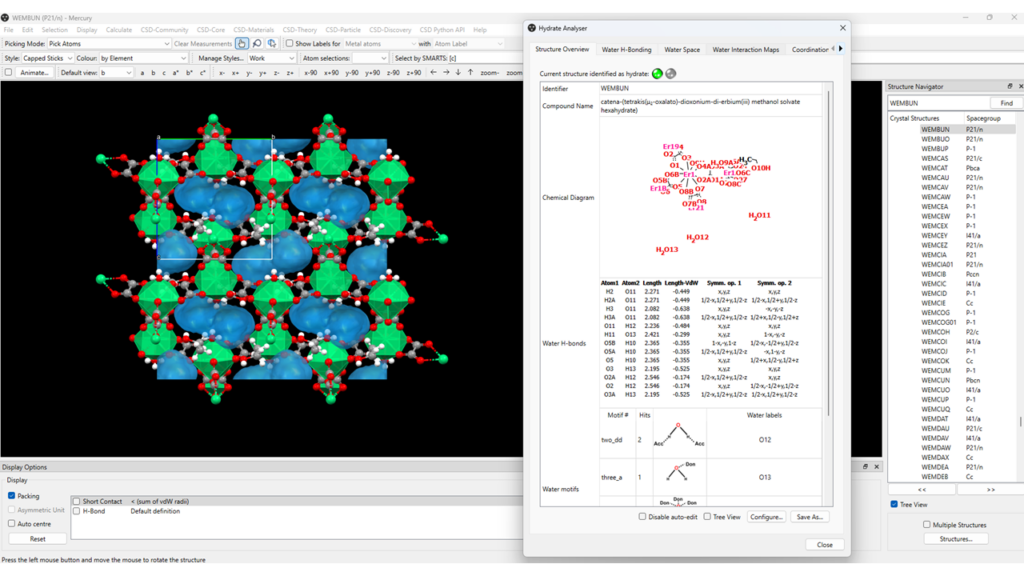
Similarly, when one or more solvents other than water are present, you can run the Solvate Analyser.
- In our example, to calculate the volume of space occupied by the molecules of ethanol and to visualize the solvent space in the structure of the framework go to “CSD-Materials” > “Solvate Analyser” > select the ethanol molecule in the structure and then click on “Add Solvent From Selected” > “Calculate Space” (Figure 5, red);
- From the “Solvate Analyser” panel you can modify the probe radius and grid spacing used for the calculation, and you can investigate the solvent hydrogen-bonding motifs, if present in the structure.
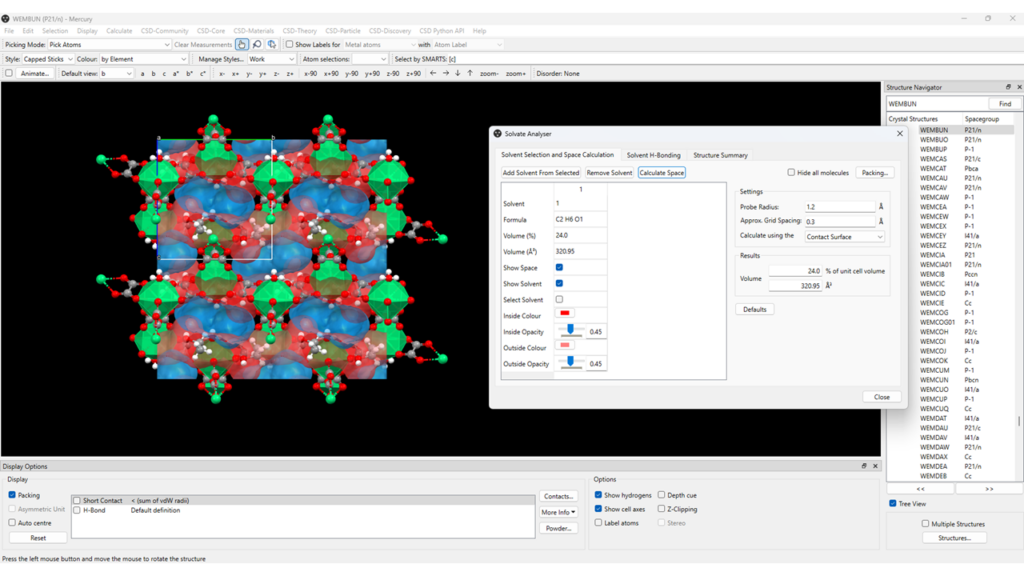
As seen in Figure 5, you can study the water space obtained from the Hydrate Analyser calculation (blue) and solvent space obtained from the Solvate Analyser calculation (red) simultaneously. However, if you prefer to analyse them separately, click on “Reset” at the bottom left of the Mercury window and then run the Solvate Analyser as indicated above.
Powder X-ray Diffraction Analysis
Finally, Mercury enables you to simulate powder X-ray diffraction (PXRD) patterns from single crystal X-ray diffraction data from the CSD or your own experimental results. This is particularly helpful to characterize newly synthesized materials and to validate experimental results.
To simulate a PXRD pattern:
- Go to “Calculate” > “Powder Pattern” (Figure 5);
- By clicking on “Customise”, a series of customizable settings are available for the pattern calculation, as well as display settings;
- The simulated PXRD pattern can be saved as an image; other formats are available as well for you to export the data (.xy, .tsv, etc).
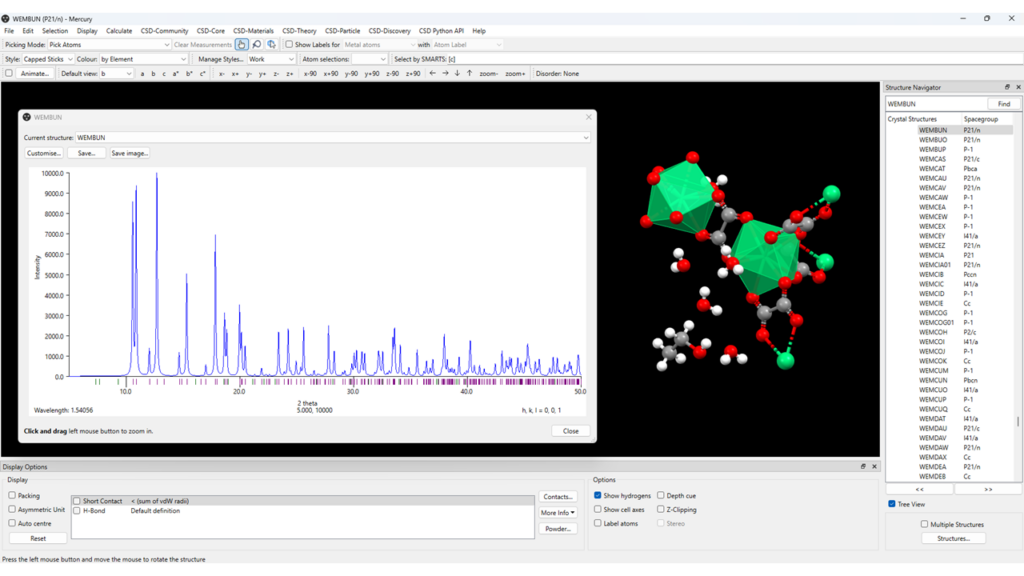
It should be noted that Mercury simulates the PXRD pattern for the selected structure, which in this case is the solvate structure, CSD Entry WEMBUN. If you want to calculate and analyse the PXRD pattern for the structure with no solvent molecules, you have to remove them from the framework (as explained above), save the modified CIF, and then load it. You’ll then be able to look at the simulated PXRD patterns of the framework with and without solvents by just selecting the files on the menu on the right-hand side.
Conclusions
In this blog we learnt how to search for MOF structures in the CSD and explored ways to visualize structural features and analyse the pores of MOF materials.
Altogether, the functionalities available with CSD-Frameworks herein presented enable efficient understanding of structure-property relationships in MOFs and accelerate the design and development of new functional porous materials.
Next Steps
- Read our white paper Accelerating Porous Materials Design and Development—A Data-Driven Approach on data, software, and a comprehensive workflow for advanced porous materials research.
- To discuss further and/or request a demo with one of our scientists, please contact us via this form or .