How to Compare Polymorphic Structures Using Mercury
Here we will demonstrate how to perform an in depth comparison of polymorphic structures using the informatics tools included in CSD-Materials.
You will learn:
- What tools are available in the CSD-Materials suite and how they can be used to compare solid forms;
- How the use of informatics tools can be beneficial to compare your polymorphic structures;
- How scientists can have greater confidence in progressing a solid form in development by combining experimental and informatics results.
Why Should You Study Your Polymorphs? How Can We Help?
Comparing your polymorphs against the variety of structural data in the Cambridge Structural Database (CSD) can help you understand which features of the structure are “unusual” and which may have an impact on the relative stability of the solid forms.
Besides containing over 1.25 million structures, the CSD also includes over 28 million bond lengths, 40 million valence angles, 14 million torsion angles and more than 2 million hydrogen bonds, representing a rich and extensive source of structural data.
The comparison of polymorphs can also help you to identify the differences between the solid forms and may highlight areas to explore experimentally.
The CSD-Materials and CSD-Core suites include structural informatics tools that you can use to study your polymorphs. These are:
- Molecule Overlay;
- Mogul Geometry Check;
- Crystal Packing Similarity;
- Hydrogen Bond Statistics;
- Full Interaction Maps (FIMs);
- Hydrogen Bond Propensities;
- Aromatics Analyser.
How to Assess Weaknesses and Strengths in Polymorphic Structures
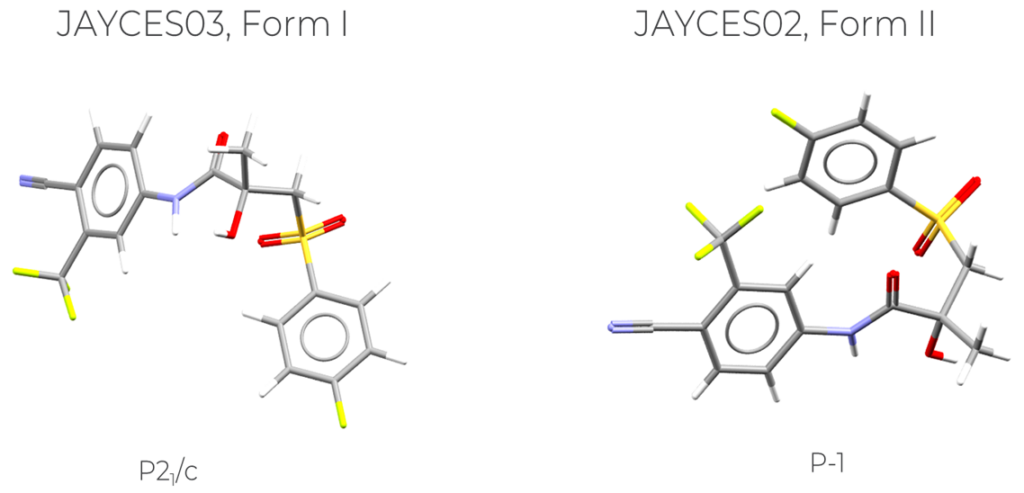
The molecules that are investigated here are two polymorphs of bicalutamide (Figure 1), an antiandrogen medication to treat prostate cancer.
The Molecular Conformations
The first step for analyzing polymorphs is to explore their molecular conformations.
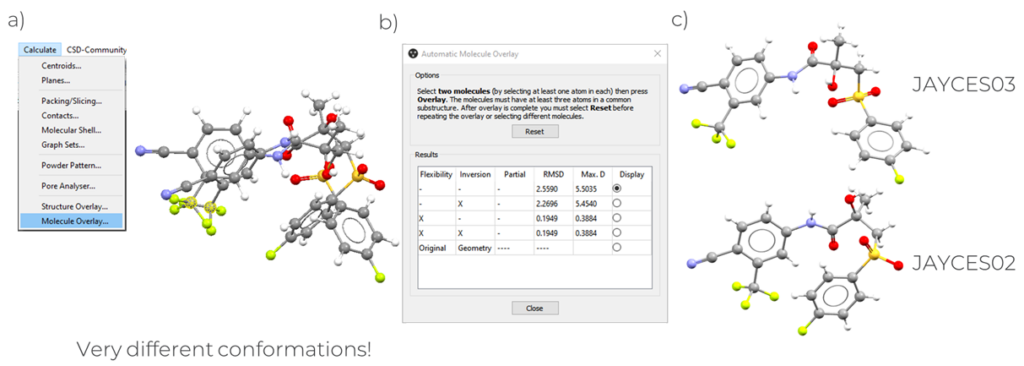
The structures of JAYCES03 and JAYCES02 (Figure 2, right) present very different conformations, and this can be seen by overlaying the two structures (Figure 2, left).
A quantitative measure of the difference between the structures of two polymorphs can be calculated using the Molecule Overlay tool in Mercury, which calculates the best RMSD overlay between the two molecules.
To do this:
- Open Mercury and search for one of the structures that you want to overlay with the “Structure Navigator”;
- Select “Multiple Structures” at the bottom of the “Structure Navigator” panel and then search for the second structure;
- Select two atoms in the two molecules on the screen and then select “Calculate” > “Molecule Overlay” > “Overlay” (Figure 2).
In Figure 2 it can be seen that the value for the best RMSD overlay obtained for JAYCES03 and JAYCES02 is high (2.56), confirming that their molecular conformations are quite different.
You can explore the conformations of your molecule in more detail using Mogul Geometry Check. The tool compares fragments of the target molecule to similar fragments in the CSD to tell you about expected values for bonds, angles and torsion angles. This allows you to quickly identify geometric features in your structure that are unusual, parts of the molecule which have the potential to be flexible, and to gain insights into the risk of polymorphism.
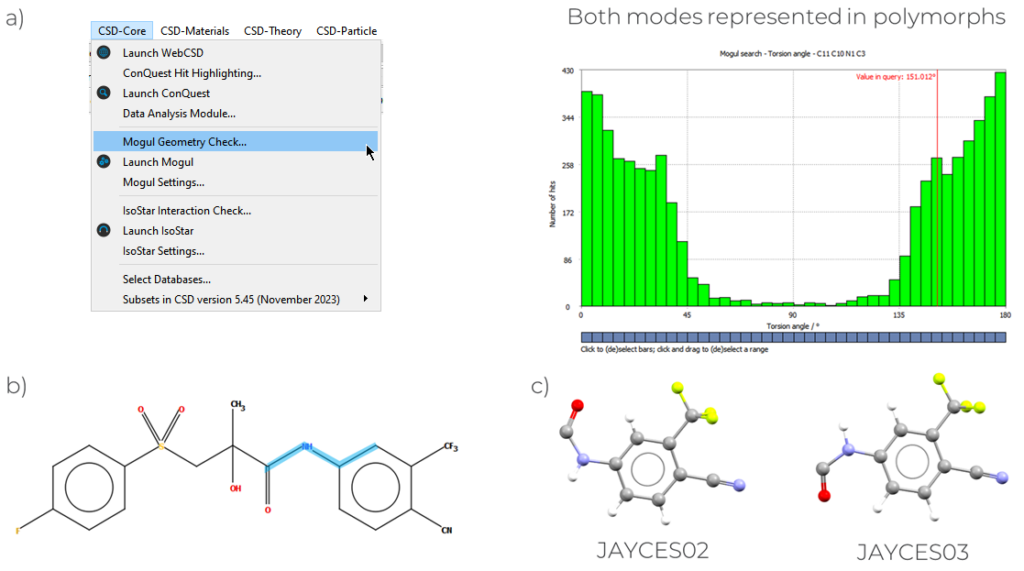
This analysis can be done from the CSD-Core menu of Mercury (Figure 3), and in the case presented here, we focus on torsion angles.
To perform a Mogul Geometry Check on a specific torsion:
- Open Mercury and search for the structure that you want to analyse;
- Select the four atoms that form the torsion angle you want to check;
- Go to “CSD-Core” > “Mogul Geometry Check” > “Search” > Double click on the torsion angle in the spreadsheet, and an histogram similar to the one reported in Figure 3 will appear;
- If you want to check the whole molecule then don’t select the four atoms in step 2.
The results from the Mogul Geometry Check (Figure 3), for the specific torsion angle highlighted in the 2D diagram, show that the fragment usually presents a torsion angle that is around 0° or 180°, and either conformation is equally likely. By performing this check on both the polymorphs it can be seen that both these modes are present, with a torsion close to 180° for JAYCES03, and 0° for JAYCES02. When the same checks were performed on the overall molecule, no torsions were classified as unusual in either polymorph.
From these analyses it can be concluded that the conformations of the two polymorphs are different, but we should not expect one to be much higher in energy than the other.
Intermolecular Interactions and Packing
Once the conformations of your polymorphs have been analysed, it is important to look at their packing environment. The CSD can also be used here, allowing you to identify unusual hydrogen bond parameters, for example, as well as enabling the comparison of structures.
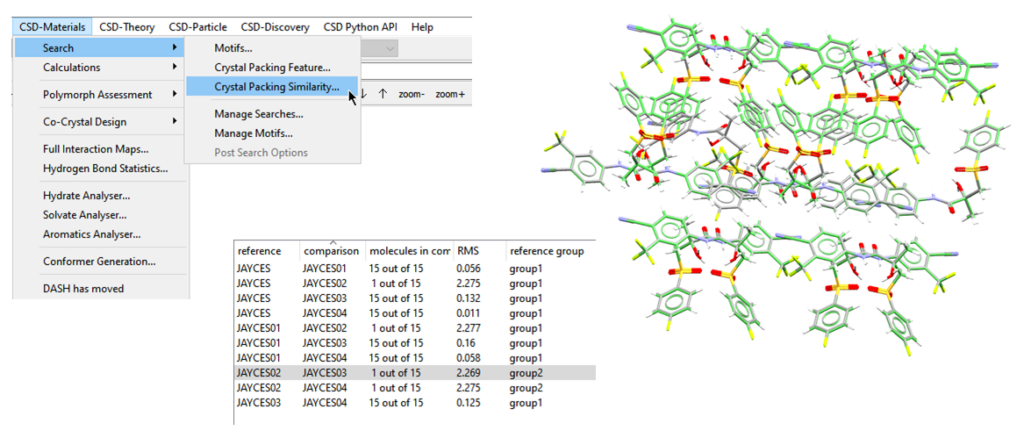
To start with, a quick step to take is just a basic comparison of the packing environments of the two polymorphs using the Crystal Packing Similarity tool (Figure 4). To do this:
- Open Mercury and go to “CSD-Materials” > “Search” > “Crystal Packing Similarity”;
- Select the Reference Structures and the Comparison Structures of your interest — in this case the JAYCES refcode family can be used for both Reference and Comparison;
- Click “Next” and change the suggested options, if of interest;
- When you reach the final window, click “Compare”. You’ll see an overlay of the packing environments of the two structures, alongside the results of the search which include the number of molecules that the two structures have in common (Figure 4).
The search can also be performed including multiple structures that belong to the same family of refcodes as they are polymorphs or redeterminations of the same crystal structure under different conditions.
The results of this analysis show that the two structures JAYCES03 and JAYCES02 only have one molecule in common out of 15, as can be seen in Figure 4, and hence their packing differs considerably.
Continuing with the structural study of our polymorphs, an important analysis that should be undertaken is the investigation of the hydrogen bond interactions in the structures.
Hydrogen Bond Statistics is a tool that allows you to use standardized functional group definitions to search for similar hydrogen bonds in the CSD, and to compare the geometrical parameters of your polymorphs with those of other deposited structures.
To perform this analysis:
- Open Mercury and search for the structure that you want to analyse;
- Go to “CSD-Materials” > “Hydrogen Bond Statistics” > “Search”;
- Expand the window obtained from the calculation, and select the different bonds listed to obtain the histograms and the heat map that can be seen in Figure 5.
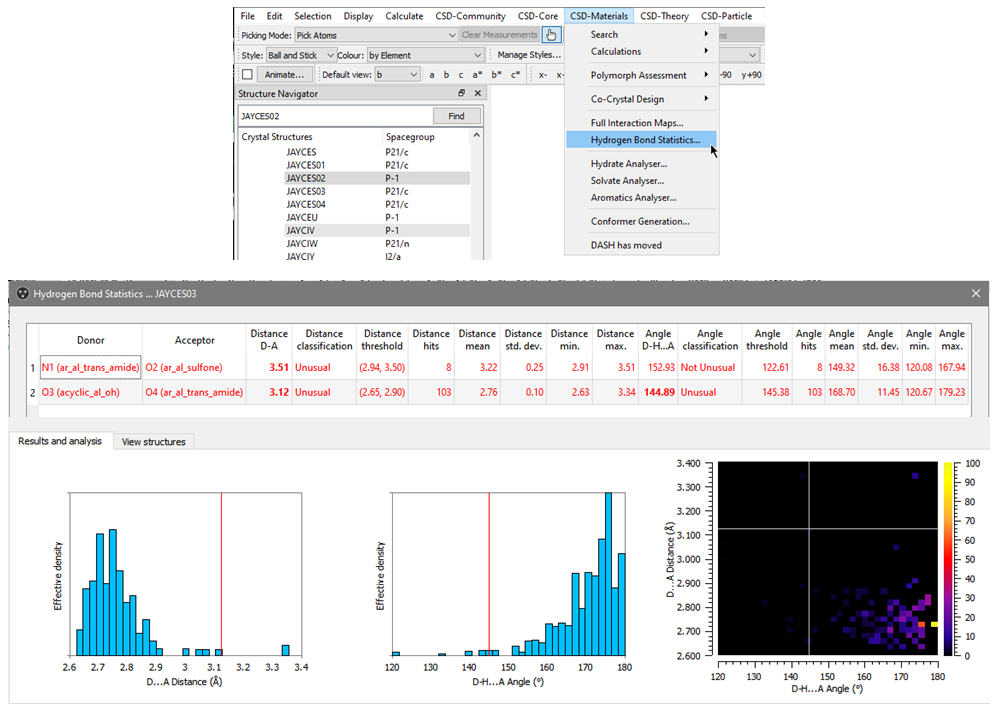
From the outputs obtained using Hydrogen Bond Statistics in JAYCES03 (Figure 5), it can be seen that both the hydrogen bonds present an unusual Donor (D) – Acceptor (A) distance and that one of them reports an unusual D-H…A angle. It should be noted that unusual geometries may suggest less stabilizing interactions.
The same analysis performed on JAYCES02 showed that the polymorphs have an amide to sulfone interaction in common (though it is an unusual interaction in the CSD), but that the second hydrogen bond, involving the hydroxy donor, uses a different acceptor in JAYCES02 from the amide chosen in JAYCES03. Neither of the hydrogen bonds found in JAYCES02 were classified as unusual. This shows that the two polymorphs have different hydrogen bond interactions, and that those in JAYCES03 might be less stabilizing than those in JAYCES02.
Full Interaction Maps (FIMs)
Full Interaction Maps (FIMs) is a visual tool that allows you to see what an ideal packing environment should provide to your target molecule. It shows density maps for where functional groups are expected to be found by breaking the target molecule into “central” groups and investigating interactions with “probe” groups to map where the interactions are expected.
To perform this analysis:
- Open Mercury and search for the structure that you want to analyse;
- Go to “CSD-Materials” > “Full Interaction Maps” > Select the probes that you want to display and the preferred options for the Map Contour levels > “Calculate Maps”;
- It might be useful for you to display the Short Contacts in your molecule to easily compare its contacts with the calculated maps.
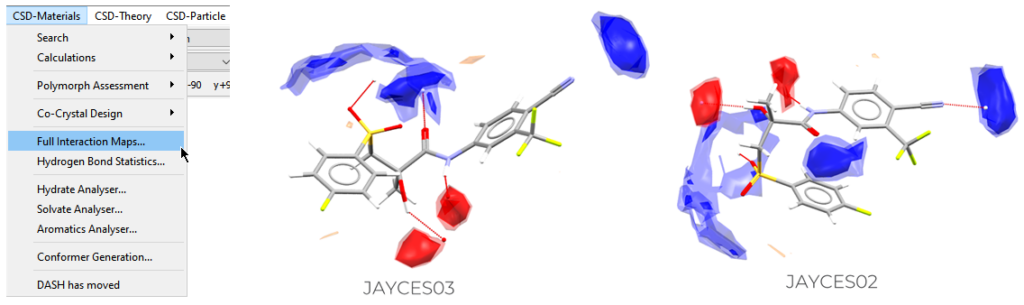
The FIMs analysis was performed on the two polymorphs of bicalutamide.
One should expect to find acceptors in the red clouds and donors in the blue ones, but as shown in Figure 6, for JAYCES03 there isn’t a good match between the interactions seen in the structure and the FIMs plots. This mirrors what emerged from the Hydrogen Bond Statistics for unusual D-H…A geometries.
A better match can be seen for JAYCES02, but several donors are missing where interactions are expected to be found.
It can be concluded that the geometry of hydrogen bonds seems to be poor in both the structures, and an ideal packing environment isn’t found in either polymorph.
Hydrogen Bond Propensity (HBP)
Another tool that analyses the hydrogen bonds in a structure is Hydrogen Bond Propensity (HBP), which looks at how donor and acceptor groups are paired up together in a structure, and how the functional groups behave compared to those found in relevant structures in the CSD.
To perform this analysis:
- Open Mercury and search for the structure that you want to analyse;
- Go to “CSD-Materials” > “Polymorph Assessment” > “Hydrogen Bond Propensities”;
- Click “Next” > “Generate” > “Analyse” > “Fit Model” > “Accept & Calculate”;
- To get a deeper understanding of what each of these steps do, read our blog on the Hydrogen Bond Propensities tool and find out important features of HBP, including how to plot polymorphic families in the same landscape.
One of the outputs from the HBP analysis is a landscape of possible networks that are available to your target molecule (Figure 7): structures with good Donor-Acceptor pairings, and which use the functional groups appropriately, can be found at the bottom right of the landscape, while structures with low ranking are found at the top left.
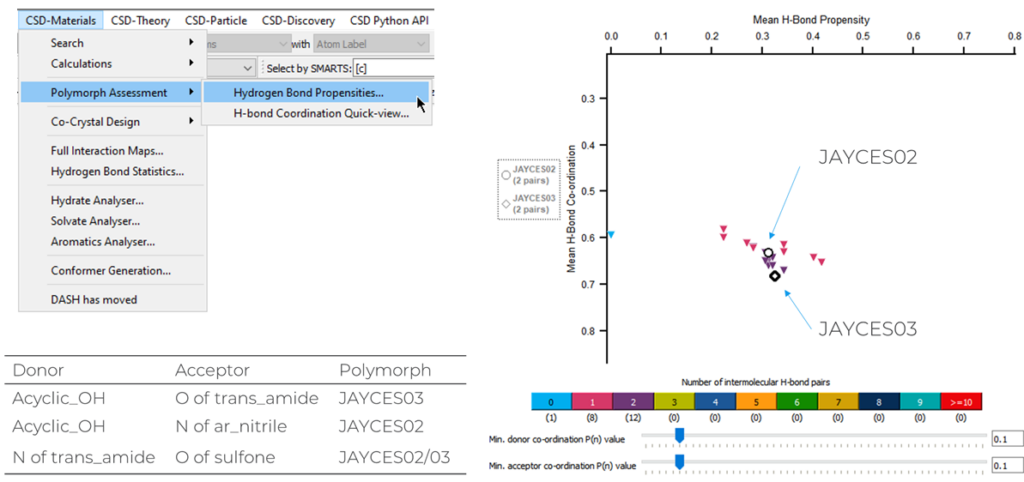
Looking at the HBP landscape reported above, JAYCES03 seems to have better use of the functional groups in the molecule and slightly better Donor-Acceptor pairings. However, both the polymorphs exhibited relatively low mean H-bond propensities, and the lowest propensities were seen for the trans-amide to sulfone interaction which was identified as unusual in the previous section.
Overall it can be concluded that the hydrogen bonds available to these structures are not ranked very highly which may suggest that multicomponent forms such as cocrystals or solvates are possible.
Aromatic Interactions
Assessing the aromatic ring interactions in a structure can give additional insights on its packing environment. The Aromatic Analyser tool can be used for this purpose, and allows you to assess the strength of interactions between C6 rings. The tool is based on a neural network that has learned the relationships between ring-pair geometries and energies of the interactions.
To perform this analysis:
- Open Mercury and search for the structure that you want to analyse;
- Go to “CSD-Materials” > “Aromatic Analyser”;
- Select the whole molecule > “Calculate”.
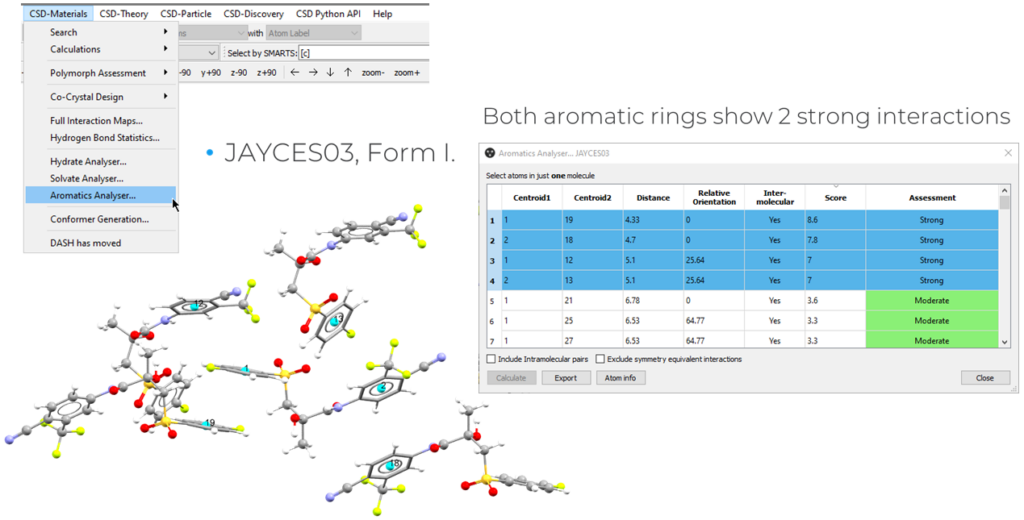
Applying this tool to both the polymorphs, it was possible to identify that JAYCES02 presented two strong intermolecular and one intramolecular aromatic interaction. In JAYCES03, instead, each C6 ring presented two strong aromatic interactions, as can be seen in Figure 8, resulting in a higher number of stabilizing ring interactions.
Conclusions
A workflow to perform an in-depth study of polymorphic structures has been reported above.
The two polymorphs of bicalutamide that we analysed here presented different conformations, but there is no suggestion that one is significantly higher than the other in energy.
The hydrogen bond interactions differ in the two polymorphs and their geometry looks more stabilizing in JAYCES02, but JAYCES03 seems to have a slightly better use of the functional groups in the molecule and better Donor-Acceptor pairings.
Overall, neither of the two structures show very good hydrogen bond propensities.
Finally, there are more aromatic interactions in JAYCES03 that may provide stability to the structure.
Overall this analysis gives us a few points to investigate:
- A lot of the analysis of the hydrogen bonding in these polymorphs comes down to where the hydroxy proton is placed and different polymorphs show different orientations. Are we happy with the hydroxyl proton position in JAYCES03?
- Due to the low propensities of hydrogen bonds, is there the potential for multicomponent forms for the system?
Next Steps
Discover more about different functionalities available in Mercury to analyse polymorphic structures and assess their stability. Watch our Virtual Workshop: In depth comparison of polymorphic structures using Mercury and find more material, including exercise handouts here.
To discuss further and/or request a demo with one of our scientists, please contact us via this form or .