Why computational chemists should follow CSP developments?
The balance between computational efficiency and accuracy is often a tricky thing to get right, and knowing which types of methods are available and what they're capable of can be difficult to summarise and keep up with. With the continuous increase in computational technologies and access to more powerful HPCs, CSP methods are providing greater insights to solid-state materials than ever before. With their applications growing (fast!), the wider computational chemistry community may want to keep an eye on CSP developments and what's next for this expanding research area.
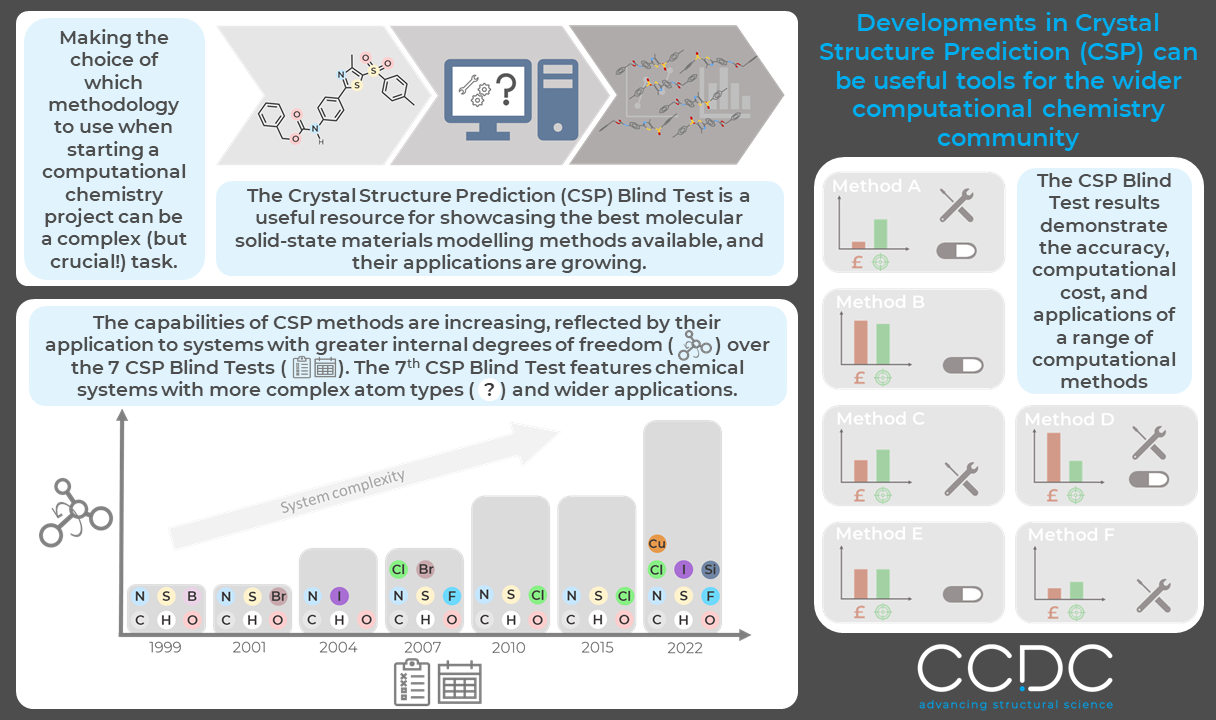
As a computational chemist, a must-have for any project is the most efficient and accurate computational method to describe the chemical system(s) of interest. Without a clear justification for your decision to use a chosen functional or particular set of parameters to describe your system, particularly for those starting a fresh project, readers may deem your results and conclusions as invalid (and justifiably so!). This leads many to carry out an extensive review of the literature to compare suitable methodologies, which often gives conflicting results, or even carry out their own method validation (which is a whole project in itself for complex systems!). The Crystal Structure Prediction (CSP) Blind Test is an extremely useful resource for this.
For those who may not know, the CSP Blind Test is an initiative coordinated by the CCDC to enable those at the forefront of CSP methods development to carry out a blind testing of their methods in an unbiased, controlled manner (the chemical systems tested are unpublished crystal structures sourced by the CCDC, overseen by an external referee, from crystallographers across academia and industry which are kept strictly confidential throughout). It may seem from the name that the CSP Blind Test serves its purpose within the CSP community where focus has historically been on assessing the risk of polymorphism in pharmaceutical materials, though taking a closer look at what CSP involves, it serves a much wider purpose. From a basic viewpoint, CSP involves two steps, first, the generation of possible solid-state configurations or crystal forms, and second, accurately ranking the generated forms in terms of stability to determine the most likely structure(s) to be observed. Both steps have a wide range of applications in computational chemistry investigations of molecular solid-state materials. In the 7th CSP Blind Test, both steps will be tested and analysed individually for all methods applied to each system. The results will then showcase the computational methods that are available (or in development), their computational expense, accuracy, and applications. An interesting development for the current Blind Test is the introduction of both Cu and Si-containing systems to the test set which aims to push the boundaries of CSP beyond the pharmaceutical sector to areas such as electronics and photonics.
With the increasing versatility of CSP methods, and their evolution from a complementary tool to a more reliably predictive one, those interested in the characterisation and design of functional molecular materials should find some exciting developments in CSP (in both the present and years to come!), and for those embarking on a new project, a go-to guide on what methods are out there in the CSP Blind Test.
More on CSP:
- Video – Introduction to Crystal Structure Prediction (10 minutes)
- Blog – will CSP be the next challenge for tech giants like Amazon or IBM?
- News – the CSP blind test begins – why it’s important, and how it compares to other challenges
- Literature – past CSP Blind Tests – summaries from previous tests and links to the publications