CSD-Materials in Action: An Integrated Approach for Solid Form Derisking
This blog highlights the recent work by Ghazala Sadiq and co-workers from Pfizer and the Cambridge Crystallographic Data Centre (CCDC), in which the structural informatics tools included in CSD-Materials were used for solid form derisking.
The paper combines experimental, informatics, and energetics methods to analyse four polymorphs of PF-06282999, a drug candidate with conformational flexibility and various possible hydrogen bond networks.
This combined approach has shown to be excellent for analysing the solid form landscape of PF-06282999 and for assessing risks associated with the solid form selection, crucial aspects that can guide scientists in making key decisions when developing a new drug product.
Why Should You Investigate a Solid Form Landscape?
Identifying the best solid form for a drug candidate is not straightforward.
A crystalline solid can present itself in the form of a salt, a hydrate, a cocrystal, a solvate. Different packing arrangements of the same molecular entities can also arise—forming the so-called polymorphs.
The crystal packing arrangement of a solid form influences both its thermodynamic properties (for example the solubility) and its kinetic attributes (such as the rate of dissolution, and the physical and chemical stability). Being able to investigate a solid form landscape is hence of paramount importance when developing a commercial drug product.
Aiming to generate structural informatics tools to support decision making in solid form design, Pfizer and other world leading biopharmaceutical companies have been collaborating with the CCDC for over a decade now through the Crystal Form Consortium (CFC).
In this white paper you can find out more about the origins of the CFC, the organizations involved, and the outputs derived from this collaboration.
Why the CCDC?
The CCDC specializes in the collation, preservation, and application of scientific structural data for materials and life sciences research and development.
The Cambridge Structural Database (CSD) is curated by the CCDC and is the world’s largest database of experimental crystal structures, currently comprising of over 1.3 million small molecule organic and metal-organic compounds.
Every structure in the database is a source of structural knowledge, containing information on the bond lengths, bond angles, and hydrogen bond interactions that are present in that specific crystal form obtained experimentally.
When the features of thousands of crystal structures are examined simultaneously, these can give valuable insights on usual or unusual intramolecular and intermolecular interactions, and can help identify high energy conformations or poor geometry.
A CSD-derived knowledge approach, based on the comparison of features of a target structure with those of relevant fragments from molecules in the CSD, is at the foundation of a digital workflow developed by the CFC members. This is called Solid Form Health Check.
What Is a Solid Form Health Check?
A Solid Form Health Check is a digital workflow that uses solid-form informatics for the assessment of small molecule drug candidates.
The workflow was first published in 2012 (CrystEngComm, 2012, 14, 2391-2403), and has been further optimized (Journal of Pharmacy and Pharmacology, 2015, 67, 857–868) and partially automated with the Python API since (J Appl Crystallogr, 2024, 57, 1235-1250).
Reported in Figure 1 is an overview of the Solid Form Health Check workflow, which includes steps to assess the risk of polymorphism, and rationalize aromatic and H-bond interactions in solid forms. The workflow can be performed through Mercury.
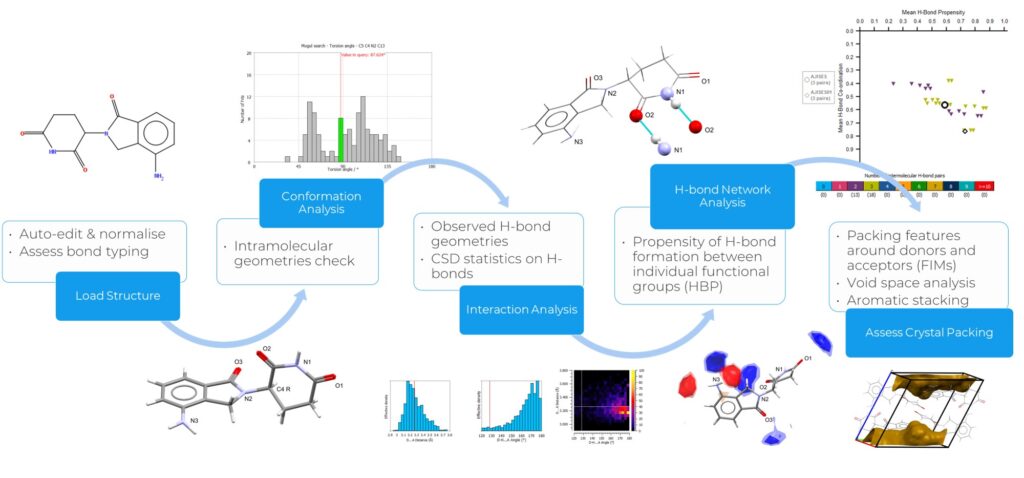
In the work herein presented, the researchers combined this workflow with experimental and energetics methods to analyse potential risks related to the physical stability of PF-06282999, a myeloperoxidase (MPO) inhibitor.
How Was the Solid Form Analysis Performed?
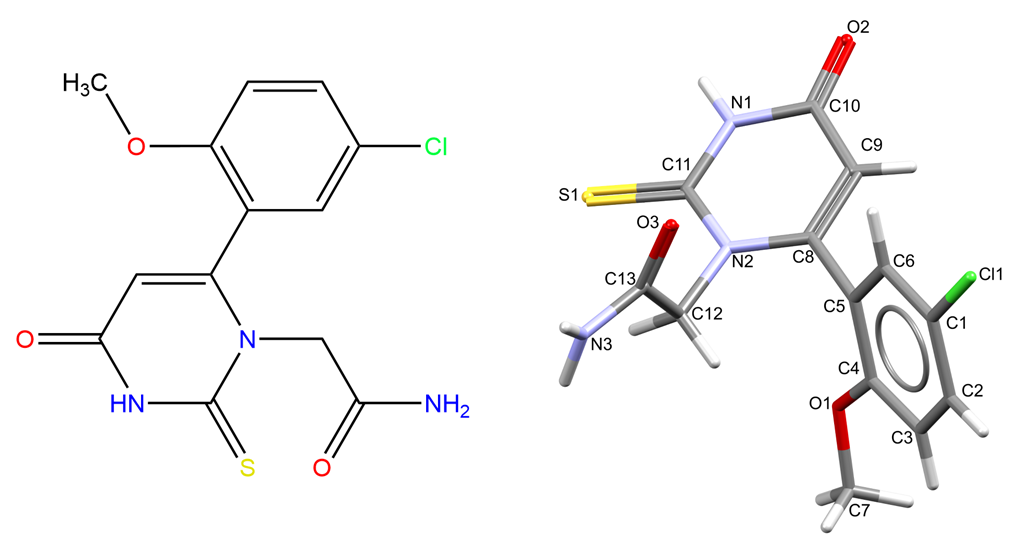
PF-06282999 presents four rotatable bonds (O1-C4, C5-C8, N2-C12, C12-C13), two hydrogen bond donors (N3, N1), and five hydrogen bond acceptors (Cl1, S1, O1, O2, O3, Figure 2). Different conformations and donor-acceptor combinations are hence accessible in the solid state for PF-06282999.
Four crystalline polymorphs of PF-06282999 were obtained experimentally, named Forms 1‒4, and the Solid Form Health Check workflow was applied to each of these. Only the most relevant features of the four polymorphs will now be highlighted: for more information, follow the link to access the full article.
After investigating the differences in conformations and crystal packing between the four forms, the team analysed the first anhydrous form that was obtained experimentally in early development: Form 1.
Analysis of Form 1
No unusual geometric parameters were detected in Form 1 using Mogul, and no unusual distances in any of the hydrogen bonds were found.
The Hydrogen Bond Propensities (HBP) component, which qualitatively assesses the usage and combination of donors and acceptors, didn’t show any major weaknesses in the structure. Form 1 contains some of the hydrogen bond pairs with the highest propensities (N3-O2, N3-O3, N1-O2). However, HBP flagged a deviation from optimal coordination for O2, which accepts twice in a bifurcated arrangement rather than once, and for S1, which appears to be an acceptor with high propensity but is unused in Form 1.
The hydrogen bond landscape obtained with the HBP component showed the possible hydrogen bond networks derived by different combinations of donors/acceptors. It was found that Form 1 exhibits a highly favourable network, but many others with similar mean propensity were present in the landscape.
Although Form 1 seemed to have lots of features suggesting it is a low energy form, the observations derived from this assessment indicated that other solid forms with good alternative networks were available for PF-06282999 and that further experimental investigation was needed.
Analysis of Forms 2‒4
Forms 2-4 were obtained experimentally after the discovery of Form 1.
The O2 bifurcation arrangement seen in Form 1 is avoided in Form 2, which on the other hand uses an acceptor with lower propensity (Cl1). This form also exhibited two moderately strong aromatic interactions, the highest number of potentially stabilizing aromatic interactions among the four solid forms.
Looking at the coordination likelihoods, molecule 1 of Form 3 (the only form that has two symmetry-inequivalent molecules of API in the asymmetric unit) presents an unused donor proton on N3 which appears to be very undesirable. This was also supported by the Full Interaction Maps (FIMs) analysis that showed a large, unfilled density cloud around N3 of molecule 1. However, looking at the packing it can be seen that the unused donor proton on N3 is effectively blocked from forming a hydrogen bond by the aromatic ring of an adjacent molecule.
Despite exhibiting the same hydrogen bonding of Form 1, Form 4 is considerably less well packed and only showed one moderate aromatic interaction, presenting less stabilizing aromatic interactions than the other forms. Additionally, void analyses showed that while Forms 1 and 2 have no void volumes, Form 3 has small pocket voids corresponding to 0.7 % of the unit cell volume, and Form 4 presents larger voids corresponding to 8.5 % of the unit cell volume.
Hence, solid-form informatics seemed to suggest that Forms 1 and 2 are likely to be stable, while Forms 3 and 4 present some unusual features that should be considered when looking at competing polymorphs.
Energetics Analysis
To complement the results obtained from the Solid Form Health Check workflow, energy-based methods were used to study Forms 1‒4.
An in-depth energetics analysis revealed that the lattice energies calculated for Form 4 are 10‒12 kJ/mol higher than those of Forms 1‒3, confirming the observations derived from the informatics workflow on the potential lower stability of Form 4.
Forms 1‒3 revealed to be close in energy, suggesting that more studies need to be done to further investigate their relative stability.
Conclusions
The potential risks related to the physical stability of PF-06282999 were here investigated by combining experimental, solid form informatics, and energetics methods.
Overall, Forms 1‒3 seemed to exhibit good hydrogen bonding networks and have similar energetics, while the less packed Form 4 was significantly higher in energy.
This work showed the importance of using a combined approach when analysing polymorphs, and demonstrated how structural informatics tools can help scientists to get an in-depth understanding of their solid forms.
Next Steps
Access the full article here: An integrated approach combining experimental, informatics and energetic methods for solid form derisking of PF-06282999, Sadiq, Ghazala et al., Journal of Pharmaceutical Sciences, DOI: 10.1016/j.xphs.2024.10.013.
To discuss further and/or request a demo with one of our scientists, please contact us via this form or .