How to use SMARTS and SMILES in Mercury and the CSD Python API
Note: this post was originally published 6th December 2021, and has been updated to reflect new developments in these features.
Here we look at how you can use SMARTS and SMILES in Mercury and the CSD Python API to perform substructure searches, and generate 3D molecules from strings to support your cheminformatics work. Using SMARTS and SMILES allows you to automate large numbers of queries, or perform complex searches that may not be possible by other methods.
What are SMILES in chemistry?
SMILES stands for Simplified Molecular Input Line Entry Specification. They are a way to represent a molecule in a compact way, readable by both machines and humans. They are a standard language used in chemistry and cheminformatics to represent molecular structures.
Daylight have definitions, examples, and tutorials on SMILES here.
What are SMARTS in chemistry?
SMARTS stands for SMiles ARbitrary Target Specification. They are like SMILES, but can be used to express substructures and patterns. They are the standard query language in cheminformatics toolkits such as RDKit.
For example C1CCC[*]C1 would represent any saturated, 6-membered ring containing 5 carbons and an “any” atom, as sketched below.
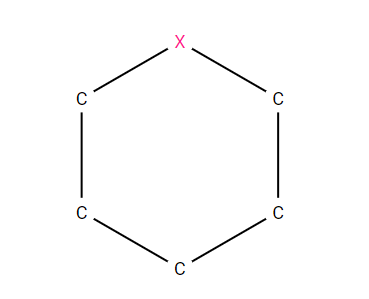 A 6-membered, saturated ring containing 5 carbons and an “any” atom, described by the SMARTS string; C1CCC[*]C1
A 6-membered, saturated ring containing 5 carbons and an “any” atom, described by the SMARTS string; C1CCC[*]C1
Daylight have a range of definitions, examples and tutorials to learn more about SMARTS.
How to use SMARTS and SMILES in Mercury
There are several ways that SMARTS and SMILES can be used in the desktop software Mercury:
- Select by SMARTS – use this search in Mercury to identify which atoms in a structure meet the search criteria. For example, searching refcode AABHTZ for the string; [C;D3!R,x2H1] returns 3 atoms, shown in the image below, which are aliphatic carbons, and are bonded to 3 non-hydrogen atoms and are not members of a ring, or have two ring bonds and one hydrogen attached.
- SMILES to 3D molecule – generate a 3D molecule from a SMILES string, with its conformation informed by the empirical data in the CSD. Find this function under the “file” menu. Learn more about this functionality here.
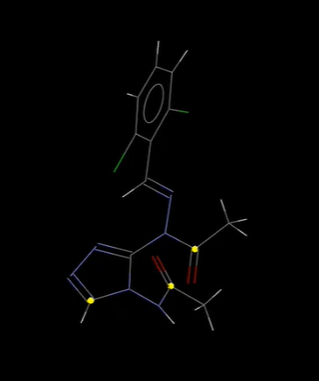
Select by SMARTS function used in Mercury to highlight atoms meeting a certain search criteria.
How to use SMARTS and SMILES in the CSD Python API
- Generate SMILES string for a molecule – quickly write a SMILES string for a molecule via the CSD Python API. This example has aromatic Boron atoms and trans double bonds.
>>> from ccdc import io
>>> io.MoleculeReader("csd").molecule("ABEHUK").components[1].to_string('smiles')
'c1cc:[B-](:cc1)/C=C/c1ccc(cc1)/C=C/[B-]1:ccccc:1'
- Substructure search – search the CSD for structures matching a specific SMARTS query. This example matches atom chirality around C7 in CSD entry AACFAZ10
>>> import ccdc.search
>>> search = ccdc.search.SubstructureSearch()
>>> search.add_substructure(ccdc.search.SMARTSSubstructure("c[C@@]1(H)OCC=C1/C=N/N"))
>>> hits = search.search()
>>> hits[0].identifier
'AACFAZ10'
- See The CSD Python API documentation for many more examples of SMARTS and SMILES uses
Using logical operators in SMARTS
In this release we have improved the handling of logic operators when working with SMARTS in Mercury – it’s now possible to use high and low priority AND statements, plus mix these with OR statements.
The logical operators used in SMARTS are;
- ! exclamation = not
- & ampersand = and (high priority)
- , comma = or
- @ at symbol = joined by an aromatic bond
- = equals symbol = joined by a double bond
<li>; semicolon=”and” (low=”” priority)<=”” li=””>
</li>;>
Using recursive SMARTS with the CSD
Recursive SMARTS allow expression of atoms that are themselves conditioned on substructures. This means that one query can encapsulate ambiguous queries, for example to return results where a group is at one position OR another.
The $ symbol is used to write recursive SMARTS. This can be used in Mercury or in the CSD Python API.
For example, if we wanted to search for methyl groups that are bound to a phenyl ring with an NH2 group meta to the methyl carbon, and an oxygen ortho to the methyl carbon. This is an ambiguous query because the NH2 group could be at the 2 or 5 position, as pictured below. This would be challenging to search for via sketches in a program like ConQuest or WebCSD.
This could be searched for in a single recursive SMARTS query; C[$(c(c[NH2])cc(OH),$(cc([NH2])c(O))]
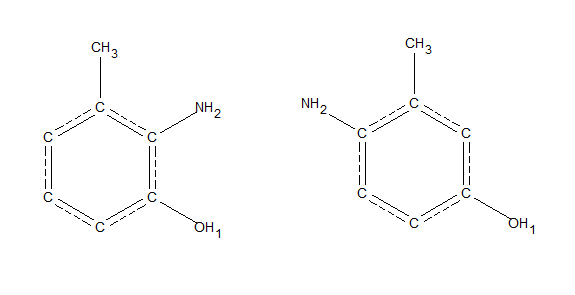
The two ambiguous structures that could be searched for with one recursive smarts query: C[$(c(c[NH2])cc(OH),$(cc([NH2])c(O))]
Recursive SMARTS can also be used to perform “not” queries, for example, to search for two aromatic rings singly bound to any acyclic atom except for a sulphur or oxygen, the query would read; c1ccccc1!@[!$([S,O])]!@c2ccccc2
Note that recursive SMARTS only ever match to one atom.
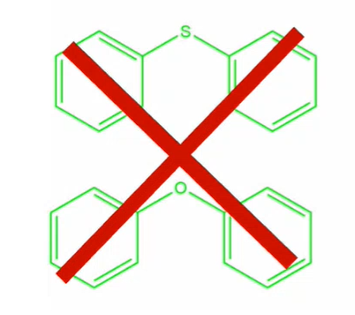
Search for two aromatic rings, singly bound to any acyclic atom except for a sulphur or oxygen – this search would be possible with recursive SMARTS string c1ccccc1!@[!$([S,O])]!@c2ccccc2
Using dot disconnect SMARTS with the CSD
Dot disconnect SMARTS allow for intramolecular and intermolecular pattern matching. They can be used in the desktop CSD program Mercury and the CSD Python API.
For example, ([Br].[NH3]) would return molecules containing both a bromine and a primary amine, which do not have to be connected.
However ([Br]).([NH3]) would return pairs of molecules containing a bromine and a primary amine, i.e. they must not be in the same molecule.
[Br].[NH3] returns hits that match either of the above cases.
Learn more
Find out more about the CSD, the Cambridge Structural Database, here.
See the latest updates to CSD software and data here.
Learn more about the desktop software Mercury here, or the CSD Python API here.