How to Generate a 3D Molecular Structure from a SMILES String
Summary
This blog post introduces how the CSD Python API now converts a SMILES string directly into a 3D molecular structure. This gives faster, more reliable ligand preparation, useful for crystallography and chemistry workflows, using experimentally informed geometry from the Cambridge Structural Database (CSD).
Top Takeaways
- You can now input SMILES strings (including stereochemistry) into the CSD Python API to generate molecules without initial coordinates.
- The CSD Conformer Generator automatically builds accurate 3D conformers using structural knowledge from 1.4M+ experimental entries.
- Generated molecules can be saved in formats like mol2, enabling smooth integration with protein–ligand docking tools such as GOLD.
- Stereochemistry can be controlled via SMILES markers and exported back to SMILES when needed.
- This functionality requires a CSD‑Discovery, CSD‑Materials, CSD‑Enterprise, or an academic licence.
Watch the webinar on demand, including a demo and Q&A around this feature.
Introduction
Computational chemists can now create 3D coordinates from SMILES in the CSD Python API. This “1D to 3D” or “SMILES input” functionality will improve ligand preparation workflows in computer-aided drug design projects. This quick approach can be used in ligand preparation for virtual screening with GOLD or any docking program. Importantly, the structures generated are based on knowledge of existing experimentally determined systems in the CSD.
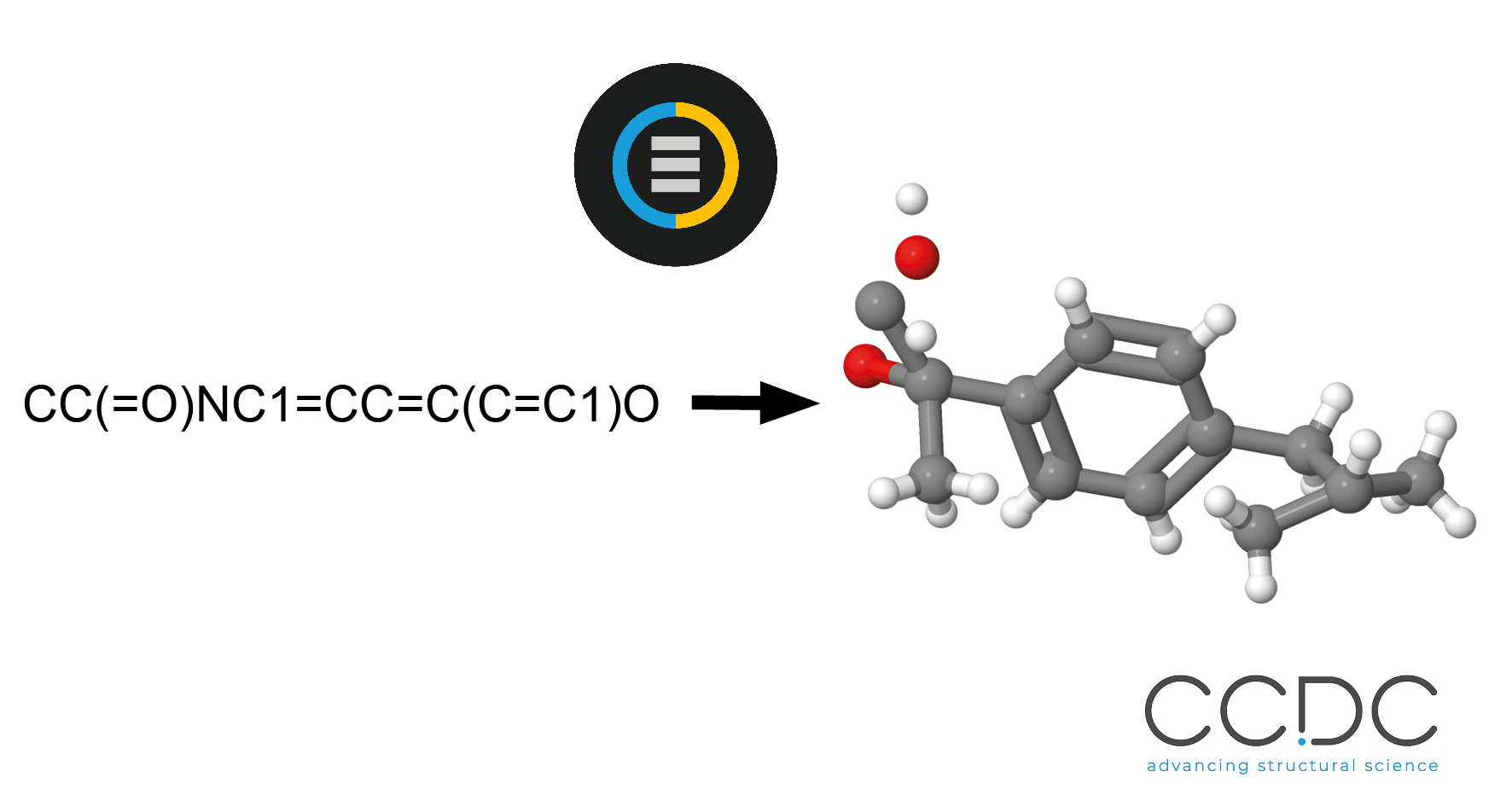
SMILES to 3D Structure Generation
The CSD Python API allows a wide range of input molecule types, such as mol2, mol and cif, as well as access to molecules within the CSD. These input formats typically provide 3D atom coordinates, which are required to initiate some workflows such as Conformer Generation and Ligand Preparation.
The CSD Python API allows SMILES strings to be used as a molecule input format, resulting in molecules without atomic coordinates, and the Conformer Generator functionality will now accept molecules and atoms without coordinates as a starting point for Conformer Generation.
The key features are:
-
Molecules can be read from SMILES strings, with stereochemistry information preserved.
-
3D conformers can be generated from such molecules and any other molecule without initial 3D coordinates.
-
SMILES strings with stereochemistry information can be generated from molecules.
How Does CSD SMILES to 3D Structure Work?
The tool uses the CSD Conformer Generator, an established and trusted part of our CSD-Discovery suite. This uses knowledge from the 1.4 million+ experimentally derived structures to predict and generate appropriate conformers, so bond lengths and angles are based on known data.
The conformer generator adds an initial 3D coordinate generation phase whenever it is given a molecule without 3D coordinates. This happens when the molecule is read without coordinates, such as from a SMILES string, and also includes cases where the molecule appears to have only 2D coordinates, for example, when it is sketched in Mercury. The 3D coordinate generator is an iterative, atom-template-based process, guided by stereochemistry information and ring positioning heuristics, with continuous optimisation based on CSD geometry distributions.
-
What files types of object are generated?
-
The process generates a CSD Python API Molecule file from the SMILES string, which could then be saved as a mol2 file.
-
-
How is the SMILES to MOL2 function accessed?
-
This feature is currently available through the CSD Python API.
-
-
What licence is required to use this?
-
A CSD-Discovery, CSD-Materials or CSD-Enterprise or academic licence is required to use the SMILES to 3D structure function.
-
-
Can I control stereochemistry during ligand preparation?
-
Yes – use stereochemistry markers on the SMILES input to generate isomeric structures.
-
How to Generate a 3D Structure from a SMILES String
In this example, we have a SMILES string for citric acid loaded into a CSD Python API Molecule object. The Molecule can then be used in further CSD Python API script, however at this stage its atoms has no coordinates.
1 >>> from ccdc.molecule import Molecule
2 >>> citric = Molecule.from_string("OC(=O)CC(O)(C(=O)O)CC(=O)O")
Some workflows require a 3D conformation of a molecule. These can be generated using the conformer generator.
1 >>> from ccdc import conformer 2 >>> conformer_generator = conformer.ConformerGenerator() 3 >>> conformers = conformer_generator.generate(citric)
Stereochemistry information in a SMILES string is used during conformer generation to produce valid conformations. SMILES strings with stereochemistry information can also be generated as follows:
1 >>> d_glucose = csd.molecule('GLUCSA')
2 >>> d_glucose.to_string('smiles')
3 'OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@@H]1O'
Learn More About CSD Python API
For more details, please check the CSD Python API documentation.
Learn more about other functions for computational drug discovery and design in CSD-Discovery.
If you don’t have a licence and are interested in trying this function, contact us here to enquire.