What Is Crystal Structure Prediction? And Why Is It So Difficult?
Crystal Structure Prediction (CSP) is a computational approach in chemistry and crystallography used to determine which crystal structures a molecule can form from its 2D structure.
For chemists and crystallographers, CSP offers powerful insight into polymorphism, helping reduce experimental risk, guide material design, and improve manufacturing outcomes.
Key Takeaways
- CSP builds 3D models and energy–density landscapes to rank likely crystal structures.
- Predicting crystal formation is difficult due to multiple conformations, packing options, and polymorphs.
- CSP can reduce costly surprises, like unexpected stable forms in pharmaceuticals (e.g., ritonavir).
- It supports innovation in materials discovery by identifying promising low-energy, functional structures.
- Advances in computation, data, and collaboration are steadily improving prediction accuracy.
CSP bridges theory and experiment, helping advance structural understanding and practical applications in modern chemistry and crystallography.
What is Crystal Structure Prediction (CSP)?
Crystal Structure Prediction (CSP) refers to the ability to identify the correct crystal structure(s) that will form from a given molecule, based on its molecular structure. Most methods use informatics and computational science techniques. The field first gained popularity in the 1980s, following statements from John Maddox on how chemists still struggle to predict crystal formation.
“One of the continuing scandals in the physical sciences is that it remains in general impossible to predict the structure of even the simplest crystalline solids from a knowledge of their chemical composition.” Maddox, J. Crystals from first principles. Nature 335, 201 (1988). Maddox – a chemist and physicist by training – served as an editor for Nature for 22 years.
Typically, a CSP analysis starts with a 2D structure of a molecule. From that, CSP researchers build a 3D molecular model, and then use advanced search techniques to generate plausible crystal structures based on free energy and packing density. Along with these 3D models, landscapes are important CSP outputs.
A landscape is a scatterplot of potential conformations that graphs free energy and density. Often, the more plausible – and therefore more likely observed – structures have low energy and high density, which is why a scatterplot can prove so helpful in identifying which potential crystal structures warrant additional experimental analyses.
Why is CSP so Hard?
There are a few key reasons CSP remains a challenge today.
- Multiple molecule conformations – even small molecules with few rotatable bonds can assume many different positions in the three-dimensional space.
- Multiple crystal packing possibilities – molecules can pack in a variety of ways to form different crystal structures, which means researchers have to sample many space groups and unit cells to predict plausible crystal packing.
- Predicted structures are not always observed – researchers can generate a landscape of different structures based on energy and density, but not all of the structures will actually form. Even though we know this, it can prove difficult to identify the real structures experimentalists can observe in the lab.
- CSP can be expensive – a single landscape can cost between $10,000 and $100,000, and some analyses can take two to three weeks, while others can take two to three months.
- Polymorphs – many compounds have more than one crystal structure, or polymorphs. Stable and metastable polymorphs form under different experimental conditions. By varying conditions – like solvent, pressure and temperature – you can produce different polymorphs; while a landscape tells you about the energy of the end product, it tells you nothing about initial conditions or the kinetics to get to that end product.
- Computational limitations – standard force-fields – and even fast quantum mechanical methods – are not always good enough to rank polymorphs. Researchers must make customised force fields for each family of problems and subsequent energy rankings to make useful predictions.
Polymorph Case Study: ROY (QAXMEH33)
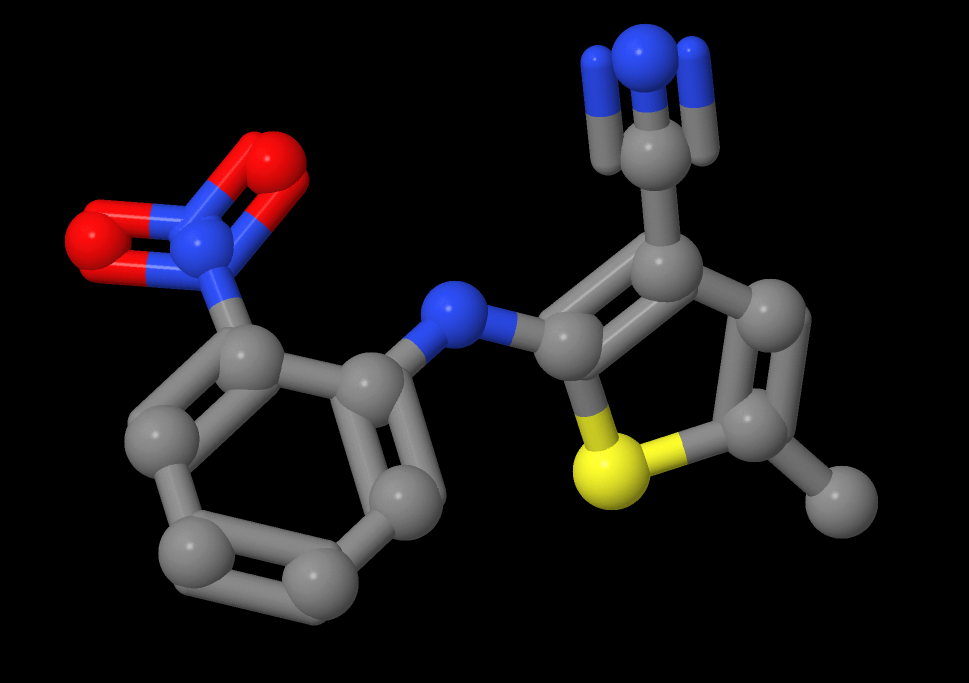
Polymorphs present a unique challenge to CSP researchers. Consider the seemingly simple molecule 5-methyl-2-[(2-nitrophenyl)amino]thiophene-3-carbonitrile – often just called “ROY” (QAXMEH33). ROY stands for “Red Orange Yellow” because the compound varies in colour based on the conformational polymorph present. (The molecule’s planarity in a given crystal determines the colour.
There are currently 13 characterised ROY polymorphs, and researchers continue to find new ones. To move forward, we likely need a deeper understanding of the crystallisation process as a whole. As Sally Price discusses in the paper “Control and prediction of the organic solid state: a challenge to theory and experiment,” we need to move from just structural prediction to predicting structural creation, which depends on experimental conditions. Read more about it here: Proc. R. Soc. A. 474(2217), 1471-2946 (2018).
What Does CSP Provide?
A CSP landscape has several applications, including:
- Risk avoidance – discovering more stable structures while establishing manufacturing processes can be costly. Computational models help identify potentially stable polymorphs ahead of time to help limit such risks.
- Manufacturing improvements – computational methods might uncover structures more conducive to manufacturing processes, and they can help determine if an observed crystal is likely to be a highly stable form.
- Patent breaking/protection – discovering crystals with better formulation or different solvation properties can cause companies to lose their patents. Companies can use CSP to look proactively for such polymorphs, to both protect their patents or to break other companies’ patents.
- New materials discovery – CSP can help researchers identify specific, potentially stable compounds with desirable qualities that they can then try to synthesise in the lab.
Risk Avoidance Case Study: Ritonavir (YIGPIO)
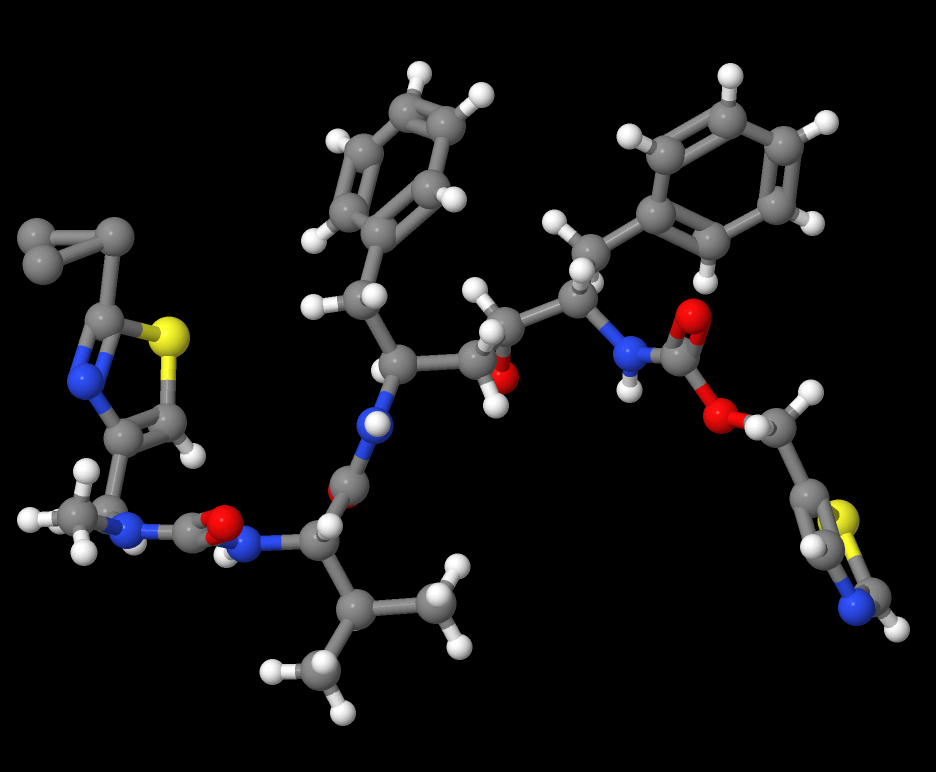
Ritonavir is an HIV antiviral and a famous example of issues caused by unexpected polymorphs. A late-occurring, more thermodynamically stable polymorph appeared during the scale-up process for manufacturing ritonavir. Once the more stable polymorph appeared, it proved difficult to produce the original therapeutic version.
It’s estimated to have cost $250 million in lost revenues, and it isn’t an isolated case. Several additional examples have occurred since, which you can read about here: Bauer, J., Spanton, S., Henry, R. et al. Ritonavir: An Extraordinary Example of Conformational Polymorphism. Pharm Res 18, 859–866 (2001) . Further studies have suggested that even existing drugs may face similar challenges. Marcus A. Neumann and Jacco van de Streek used CSP to analyze this, and they found that between 15 and 45 percent of small-molecule therapeutics are distributed using a seemingly metastable form.
“Based on a thorough and critical analysis of the commercial crystal structure prediction studies of 41 pharmaceutical compounds, we conclude that for between 15 and 45% of all small-molecule drugs currently on the market the most stable experimentally observed polymorph is not the thermodynamically most stable crystal structure and that the appearance of the latter is kinetically hindered.” Faraday Discuss., 2018,211, 441-458
CSP can help identify these types of issues before manufacturing process are put in place. Consider the (toy) landscapes below.
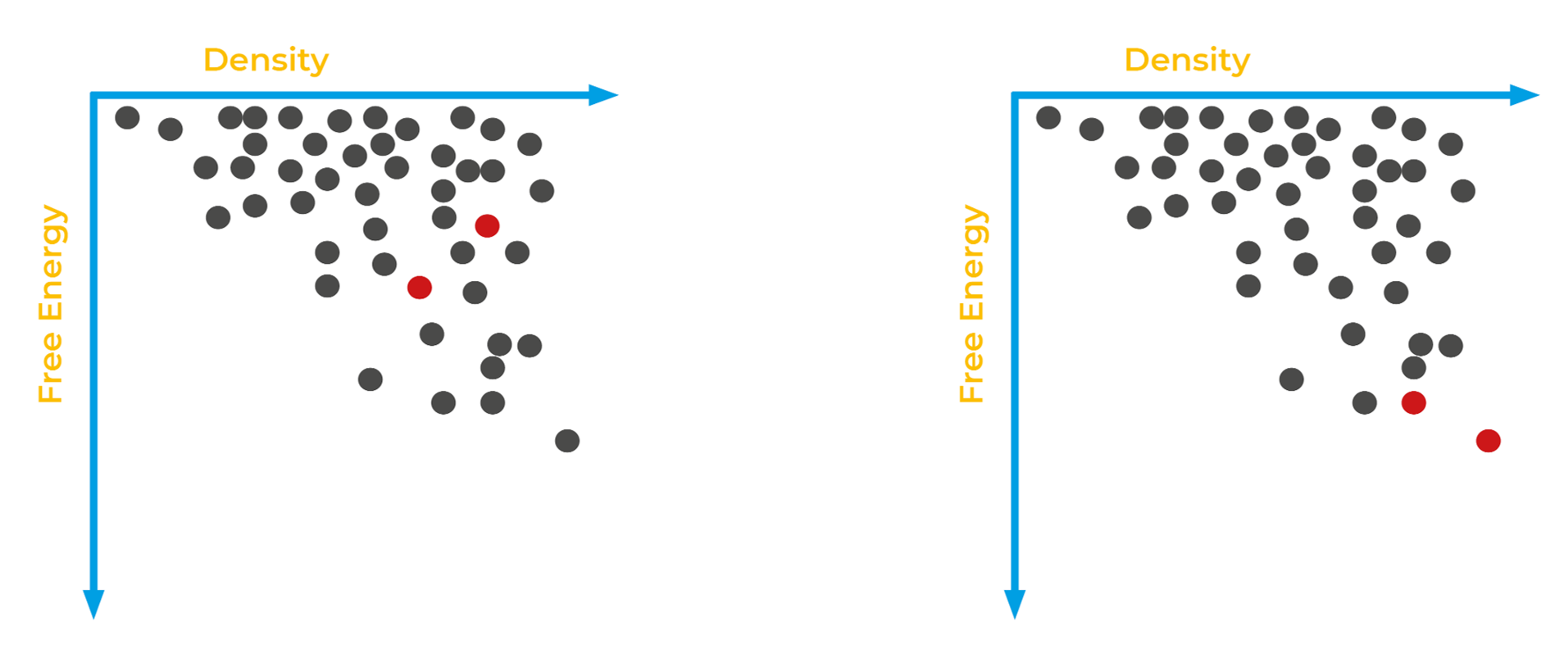
Imagine that the experimentally observed forms of your crystal are in red, while all other computationally predicted crystal forms are in grey. If your observed forms appear in the landscape on the right, then you can feel more confident that your structures are lower energy and potentially more stable. But if your observed forms appear in a landscape like the one on the left, you know additional lab research might uncover more thermodynamically stable polymorphs.
Materials Discovery Case Study: Pulido et al.
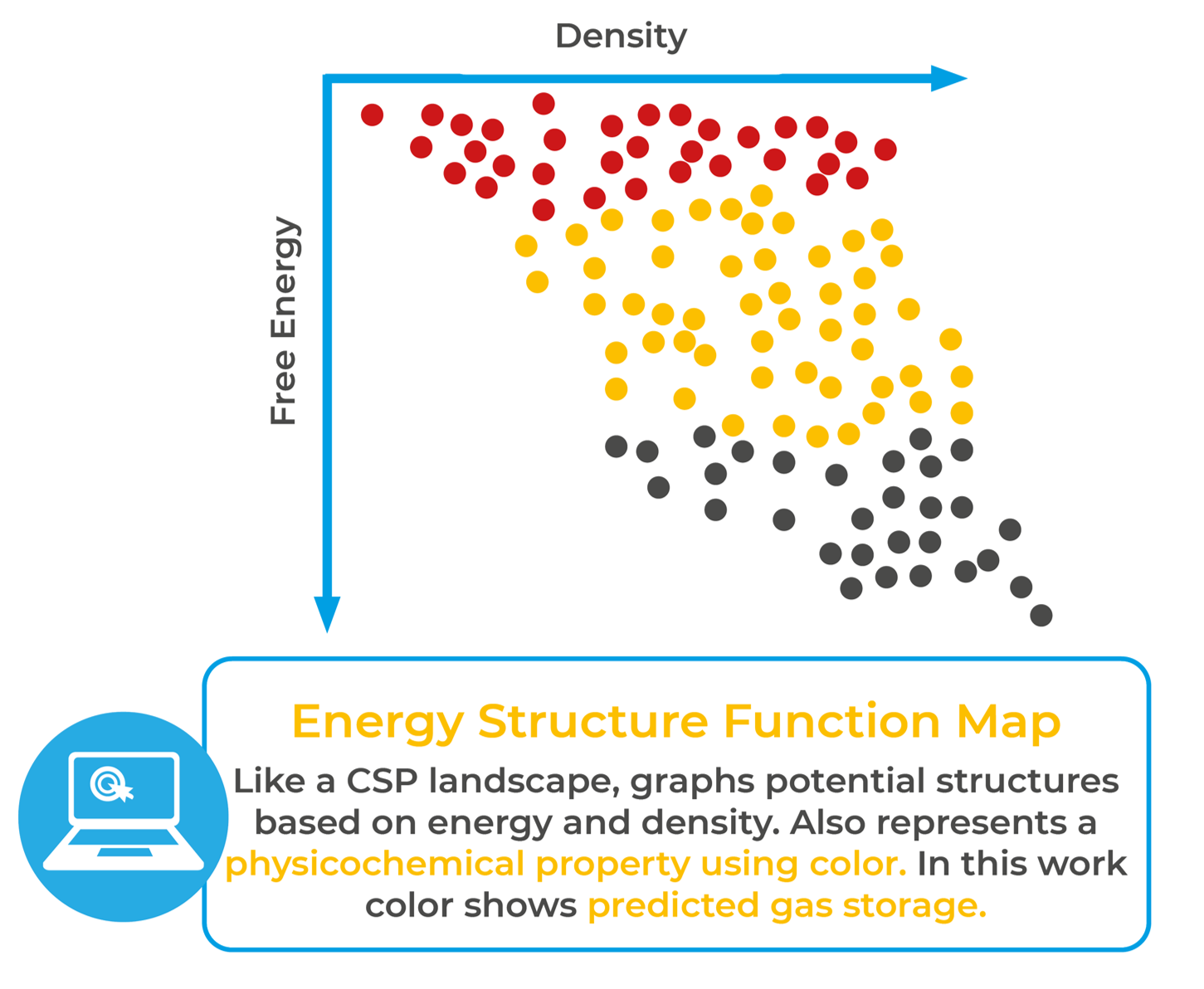
Research by Pulido and co-workers used CSP techniques and energy-structure-function (ESF) maps to search for structures with high degrees of void space that might produce compounds with applications in gas absorption. Like a CSP landscape, an ESF map graphs predicted crystal structures according to density and energy. However, it also represents a third, physicochemical property using colour – in this case, predicted gas storage. Pulido, A., Chen, L., Kaczorowski, T. et al. Functional materials discovery using energy–structure–function maps. Nature 543, 657–664 (2017).
The team produced CSP solid-form landscapes of a series of molecules and then projected the value of properties important for gas storage onto the CSP landscape. Properties like channel dimensionality of the void space and calculated methane capacity could then be viewed alongside packing and energy. This allowed the team to observe low-density structures that still appeared energetically stable, which they could attempt to synthesise in the lab. This is how ESF maps guide material design and development.
Experimentalists successfully synthesised the predicted crystal structures of a triptycene molecule (T2) and were able to observe the low-density structures in the lab. Here’s an example: DEBXIT01.
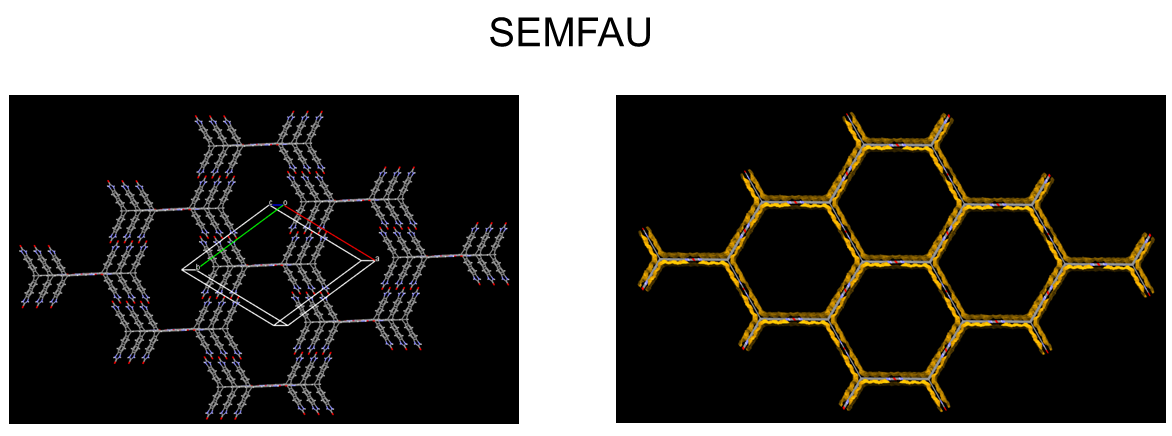
The team also investigated designing molecular crystals with even higher porosity levels with a CSP solid-form landscape of a hypothetical, extended T2 form, called T2E. They successfully predicted an ultra-low-density solid T2E molecule: SEMFAU. SEMFAU is an isostructural of the T2 form with a predicted density of just 0.303 gcm−3 and hexagonal pore channels with diameters of 2.83 nm. The team successfully synthesised it in the lab – further demonstrating how exploration of the solid form landscape of organic molecular crystals via CSP combined with physicochemical projections (ESF map) can inform functional materials design.
Where is CSP Headed?
Ideally, in the next 30 years, we’ll be able to take a very elaborate molecule and then quickly predict all its solid forms – including salts, solvates and useful conformers – without false positives on a small computational resource. We will then derive calculated information about the predicted structures, such as how to reliably make each form in the lab. We’ve still quite a way to go.
How is the CCDC Advancing CSP?
CCDC is dedicated to advancing structural science, and we sit in a unique position as a charity, as a place of active research and as producers of leading chemical information software. We can bring together academic institutions, pharma companies, other software providers and chemical data standards organisations with the common goal of advancing CSP.
The CSP Blind Test
For the past 30 years, we’ve held a blind test aimed at CSP developers in industry and academia with the goal of testing new CSP methods and demonstrating to the broader scientific community the usefulness of CSP. Each year, we work with participants all over the world.
- We canvas collaborators to provide experimentally analysed molecules that are not in the public domain.
- We release the structure with some experimental conditions.
- We give participants one year to submit their predictions using their best CSP methods.
- Once the submissions are in, we review the experimental results and compare them to the predictions.
- We review the outcomes and then publish the findings.
The event always generates lots of discussion, learning, and method improvements while giving the participants the opportunity to test and validate their techniques against real data. There are usually participants from pharma and academia who use a variety of different approaches. In the last blind test, there were five systems of differing complexity. We received 25 submissions from 52 academic groups worldwide. Each compound was predicted by at least one of the groups, and the successful approaches highlighted the importance of re-ranking using density functional theory (DFT).
The results and conclusions of the 7th CSP Blind Test have been published in Acta Crystallographica:
- The Seventh Blind Test of Crystal Structure Prediction: Structure Generation Methods – Hunnisett et al., J. Acta. Cryst. B80, Dec 2024.
- The Seventh Blind Test of Crystal Structure Prediction: Structure Ranking Methods – Hunnisett et al., J. Acta. Cryst., B80, Dec 2024.
What’s Next?
Sign up for our monthly newsletter for tips, news, and more about structural science from the CCDC.
Learn more about the CCDC’s current CSP Blind Test.
Learn more about using CSD-Theory to manage CSP data here.