CSD in Action: Using Machine Learning to Predict the Crystalline Properties of Molecular Compounds without their Crystal Structures
Here we highlight a paper by scientists at the Lawrence Livermore National Laboratory using the Cambridge Structural Database (CSD) to train models to predict the densities of molecules from chemical structure alone. This is part of our series highlighting examples of the Cambridge Crystallographic Data Centre (CCDC) tools in action by scientists around the world.
Summary
In this work, the authors demonstrate that machine learning methods that leverage quality datasets can directly learn the relationship between chemical structures and bulk crystalline properties of molecules, even in the absence of any crystal structure information or quantum mechanical (QM) calculations. The authors focused specifically on energetic materials called high explosives (HEs) and predicting their crystalline densities without crystal structure information. How to best featurize molecules as inputs into machine learning models remains a challenge within the chemistry machine learning community. The authors sought to understand whether expert-crafted features or learned molecular representations via graph-based neural network models yield the best results.
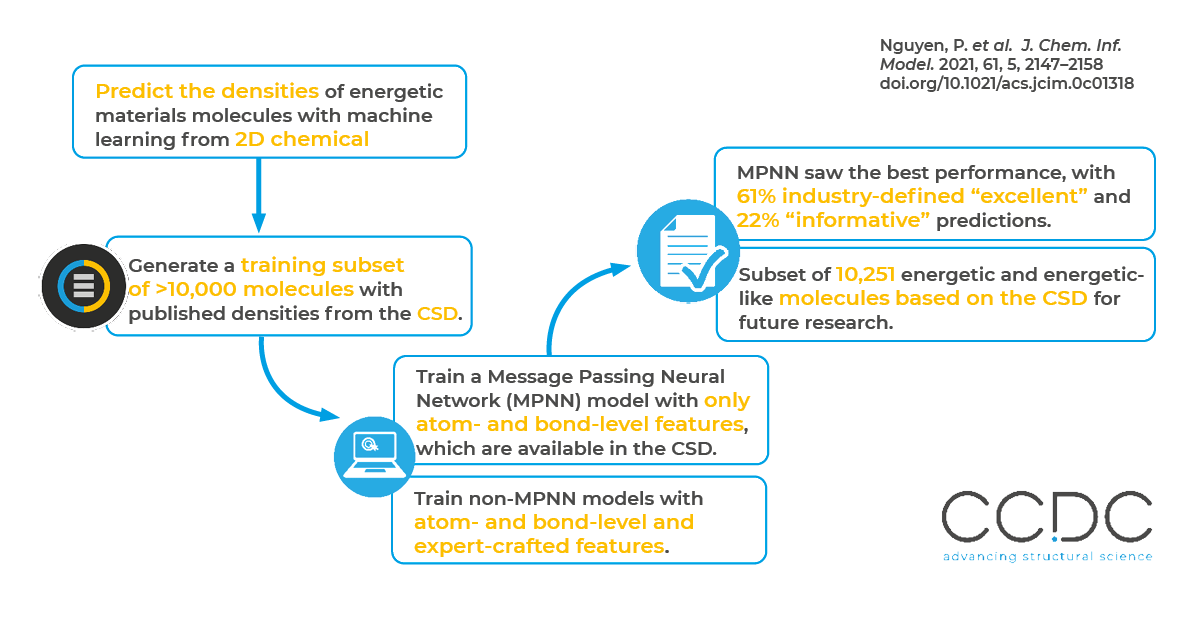
By leveraging the experimentally derived and hand-curated data in the CSD, the researchers were able to produce models that outperform current state-of-the-art methods, including more computationally expensive density functional theory (DFT)-based methods. They found that their message passing neural network (MPNN)-based models with learned molecular representations generally performed best – performing well even on data not representative of the training data. In their tests across more than 10,000 HE-like molecules, 61% of the best model’s predictions were “excellent” (error is less than 0.03 g/cm3) and 22% were “informative” (error is within 0.03 and 0.05 g/cm3) The best model also performed well predicting the densities of notoriously difficult molecules, such as riaminotrinitrobenzene (TATB) – yielding a prediction of 1.95 g/cm3, which is only 0.02 g/cm3 higher than the experimentally reported value.
Why
Predicting molecules’ bulk properties of interest from a chemical structure alone remains a long-sought-after goal in chemistry as it would expedite the discovery of novel materials and save resources on benchtop synthesis experiments across industries. A specific challenge to machine learning for HE molecules has been the lack of readily available large data sets. For example, the authors state that data sets of 300 HE molecules are considered large. Using the CSD, the researchers generated a library of 10,000 molecules with which to train their models.
How
A variety of ML approaches have been employed for predicting molecular-level properties, including energy levels and lipophilicity. However, using ML approaches to predict bulk crystalline properties is less common. (Often the goal is to find the crystalline structure.) The researchers curated a subset of molecules from the CSD, removing any molecules lacking published densities or 3D structures that could not be correctly constructed from a crystal structure file. This resulted in 10,251 energetic or energetic-like molecules. They then mapped those molecules’ quantitative molecular compound properties with features that are accessible to ML models.
For their best-performing MPNN model, the researchers only provided information about a molecule’s bonds and atoms. The MPNN is therefore developing potential features that predict crystal density. Researchers can then mine the features for new insights into what molecular features are important for advantageous crystalline densities. Historically, this training process works the other way around – with researchers providing features that may or may not be important.
Read More
Read the full paper: “Predicting Energetics Materials’ Crystalline Density from Chemical Structure by Machine Learning” J. Chem. Inf. Model. 2021 61 (5), 2147-2158
Learn more about how the components in CSD-Core provide essential search, visualisation and analysis features to deliver knowledge from the Cambridge Structural Database (CSD).
Read more tools in action.