Strengths and Limitations of Powder X-ray Diffraction
This blog is based on the article ‘Improving the accuracy of small-molecule crystal structures solved from powder X-ray diffraction data by using external sources’ published in Helvetica Chimica Acta in 2023 and dedicated to the memory of Prof. Dr. Jack Dunitz.
The paper highlights how important and widely used powder X-ray diffraction is nowadays, showing some of its weaknesses and limitations, and proposing ways to improve its accuracy.
Introduction
Among the plethora of diffraction techniques available single crystal X-ray diffraction (SCXRD) is a powerful characterization technique that has yielded more than a million crystal structures of different compounds, materials, and samples. The advantages of using SCXRD derive from the fact that the 3D diffraction pattern, and the well resolved and separated peaks that are obtained from a SCXRD experiment, allow the user to solve and refine the structures relatively easily. The software that performs structural solution and refinement is easy to use, there are tools to carry out standard structural validation reliably, making SCXRD a recognized method to perform crystal structure determination.
One disadvantage of SCXRD is the need to have a good-quality single crystal that is relatively big to perform the analysis, a requirement that is not always easy to fulfil. The use of powder X-ray diffraction (PXRD) has become extensively popular in the last three decades, becoming essential to access structural data of materials that cannot be grown as single crystals.
The success of PXRD derives from several factors. Firstly, it can be applied to a variety of materials, such as organic and inorganic compounds, but also proteins. Secondly, in some cases the quality of the data can be comparable to the one obtained from SCXRD experiments. Finally, it is a fundamental tool to analyse the bulk of the powder material, giving an averaged information of the sample rather than information related to one single crystal.
A limiting factor in the use of PXRD is the low accuracy of the obtained structures. Potential sources of errors when using this technique include the overlap of reflections deriving from broad peaks, which requires more intervention from the user to obtain structural data; and the lack of standard process to evaluate and validate a structural model. For these reasons, the presence of incomplete or inaccurate structure models from PXRD data is more frequent than from SCXRD.
Structure Solution From PXRD
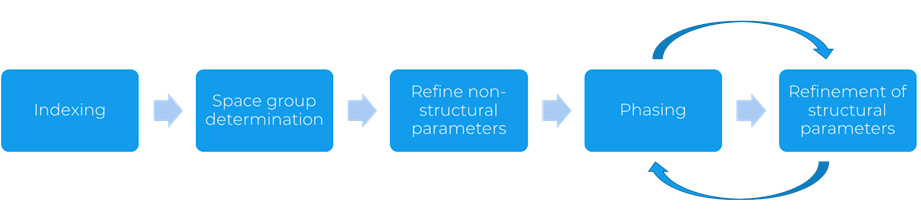
The process of structure solution from PXRD data starts with indexing, which consists in the determination of the unit cell parameters (Figure 1). The space group is then determined, and the non-structural parameters are refined. The phasing step is a crucial one, as an initial crystal structure model is produced. Finally, the atom-related parameters of the crystal structure are refined, with consequent iterative refinements between these two last steps before obtaining the final model.
This process is quite time consuming, and each step involves user input with potential user errors. The user needs to have knowledge on structure solution from PXRD to obtain a structural model effectively and reliably.
Rietveld Refinement
Rietveld refinement is one of the possible ways to perform structural refinement. It is based on least-squares optimization, which consists of minimizing the difference between the calculated and measured patterns by adjusting the peak parameters and the structural parameters. In doing so, the user adds restraints to the Rietveld refinement, such as expected bond lengths and angles with respective deviations, meaning that there is an increase in the amount of data the crystal structure is refined against.
When adding geometrical restraints in Rietveld refinement, the use of the Cambridge Structural Database (CSD) can be beneficial. The curated entries present in the database allow the determination of the median geometry parameters for the C-C bond distance in a single, double, and triple bond. These values can be used when performing Rietveld refinement to consider the expected geometries in the crystal structure.
What Can Be Done To Improve the Structure Accuracy?
This section discussed four measures that can be followed to improve the accuracy of structure determination from PXRD analysis.
Use of High-Quality PXRD Data
The use of high-quality PXRD data is one of the most important aspects to obtain a reliable structure solution, and it can lead to a model that is as good (or better) than SCXRD when synchrotron data are used.
The article reports a case in which the crystal structure of cytosine was obtained by performing unrestrained refinement (no use of geometrical restraints), owing to the small and rigid nature of the molecule, and to the high-quality PXRD data collected on a synchrotron.
A second case of a structure refined without restraints is reported for D-mannose, an example in which the unrestrained Rietveld refinement explained the incorrect structure derived from SCXRD data, caused by untreated disorder.
Complementary Analytical and Validation Techniques
Most scientists rely on complementarity of analytical and validation techniques to increase the accuracy of the structural model.
Thermogravimetric analysis can give information related to the presence of solvent molecules in the structure. The technique can give a better idea of the full chemical content of the material allowing Rietveld refinement to be performed more easily.
Infrared spectroscopy indicates which tautomer is present in the compound analysed, and to acknowledge the presence of salts or cocrystals to further help in the structure solution.
Neutron and electron diffraction techniques can be used to get additional structural information, as neutrons are better at ‘seeing’ hydrogen atoms than X-rays are, and electron diffraction can analyse crystals that are extremely small.
Nuclear magnetic resonance (1D or 2D) can be very useful to find the connectivity and the conformation of the molecules.
Computational calculations such as DFT-D and CSP can also be performed to rank the energy of different crystal structures, allowing to identify the ones that are more likely to form.
The article reports an example regarding this specific case, in which the use of complementary techniques identified the correct structure of a 1/4 hydrate cocrystal of (S)-ibuprofen and L-proline. The exclusive use of PXRD wouldn’t have been enough to get the full structural information, which was achieved by using thermogravimetric analysis and computational calculations.
Chemical Sense in Crystal Structure Solution and Refinement
The use of the CSD and CCDC software such as Mercury, can be powerful tools in the structural analysis of a molecule in terms of chemical sense.
An example reported shows how the structure of a salicylic acid/anthranilic acid cocrystal (RUTGUK/01) was originally deposited with the wrong positions for the hydrogen atoms (RUTGUK). The structure presented H-H bond distances and -COOH torsion angles that were not within the median distribution for the structures in the CSD containing salicylic acid and anthranilic acid, with hydrogen bonds that looked in close proximity when visualizing the packing in Mercury.
The correct structure (RUTGUK01) revealed to be zwitterionic, with the amino group of the anthranilic acid being protonated by one of the carboxylic groups. This discovery was possible through the visualization and analysis of the crystal packing, and the consequent use of the CSD to support the expected observations.
Escaping Local Minima and Finding Geometry Details
When performing data refinement through least-squares, the chance to reach a plausible answer that is a local minimum, and not a global minimum, is high. The parameter space should be explored by attempting lots of refinements and using random numbers to perturb the atomic coordinates in each refinement cycle.
The article reports an example of a copper-based coordination polymer solved from powder diffraction data where scientists used this approach to correctly model the organic moiety, identifying that the correct form was the hydroquinone form and not the benzoquinone one.
Conclusions
In conclusion, the authors gave a list of recommendations to reliably determine a structural model. This included the use of high-quality PXRD data, the use of complementary techniques and computational models, and the use of chemical sense when performing Rietveld refinement. They reported seven illustrative case studies that can be further explored in the main article, and highlighted the importance of the CSD and CCDC software applied to a variety of cases.
Next Steps
Find out more about the Cambridge Structural Database.
Discover the full range of CCDC software tools.
Find out more about Mercury.
To discuss further and/or request a demo with one of our scientists, please contact us via this form or .