New Functionality for the Geometry Optimization of Crystal and Molecular Structures
New functionality in Mercury and the CSD Python API for the geometry optimization of crystal and molecular structures using a range of force fields is now available.
This energy-based optimization complements our existing informatics-based tools. An interface in Mercury enables an individualized workflow for a structure of interest, and our powerful Python implementation provides a high-throughput approach to energetic minimization.
Crystal Optimiser
The lattice energy of a crystal is an important indicator of stability. An experimentally determined crystal structure may not lie in an energetic minimum of a force field model. The Crystal Optimiser relaxes the structure into the force field minima, producing a more accurate lattice energy.
The Crystal Optimiser can be run on a structure from the Cambridge Structural Database (CSD) or other in-house database, making it very simple to incorporate into a workflow. The relaxed crystal structure can be saved as either a .cif or a .mol2 file, and is added to the structure navigator in Mercury, ready for further informatics or energetics-based analysis. For example, the optimized crystal structure could be used by VisualHabit in the CSD-Particle suite to produce a predicted morphology.
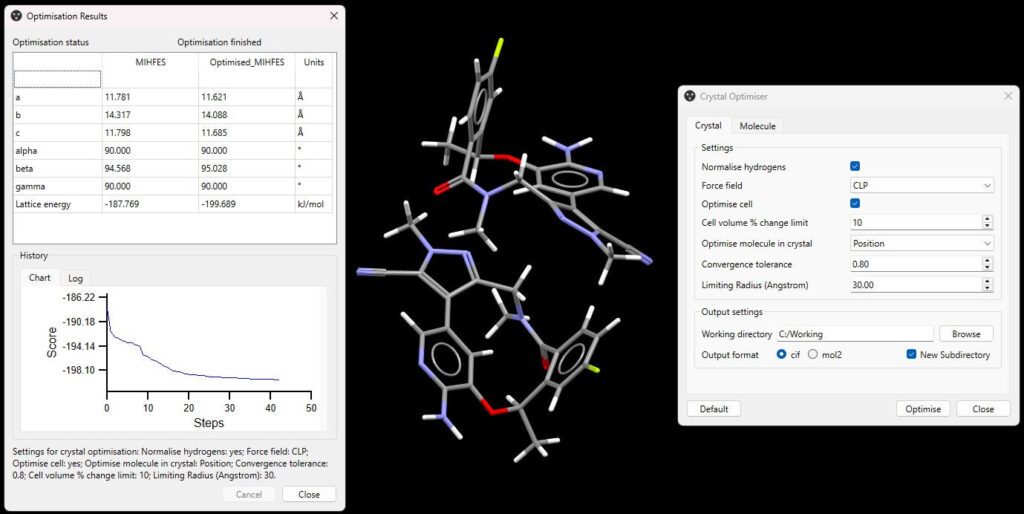
The Crystal Optimiser can use the CLP [1, 2, 3], Dreiding II [4, 5, 6], Momany [7], and Gavezzotti’s UNI force field [8, 9]. Additionally, the Cambridge Structural Database – Optimized Potential for Crystal Structures 2016 (CSD-OPCS16) force field used in the Landscape Generator [10] is made available.
The Crystal Optimiser uses these force fields to evaluate intermolecular interactions, and if needed, the Cambridge Structural Database Knowledge-Based Field (CSD-KBF) [11] can be used with these force fields to relax the intramolecular torsion angles in a combined optimization.
Molecule Optimiser
The Molecule Optimiser uses the CSD-KBF described in this paper in the Journal of Chemical Information and Modeling to optimize the intramolecular geometry of all the molecules present in a structure, producing new structures in either .mol2 or .sdf format.
The Molecule Optimiser also produces a measure of the change observed between the original molecule and the optimized molecule, as a root mean squared deviation value. As the CSD-KBF is parameterized against the Cambridge Structural Database, the Molecule Optimiser is likely to give a more crystal structure-like geometry. It provides a unique approach to molecular optimization that will be of use to scientists looking for crystal structure-like minima.
Both the Crystal and Molecule Optimisers check that the structure being assessed is ready for force field optimization (e.g., no missing hydrogen atoms) and that the force field selected is parameterized for the chemistry in the structure.
The Crystal and Molecule Optimisers are available in Mercury and the CSD Python API to CSD-Materials and CSD-Enterprise licence holders.
Developed in Partnership
The Crystal and Molecule Optimisers were developed as part of the DigiCCAMMS program, in partnership with Durham University.
This program will integrate further features from the ADDICT prototype into the CSD-Particle suite using Innovate UK funding, as part of the Sustainable Medicines Manufacturing Innovation: Collaborative R&D competition.
Next Steps
Contact us here to discuss further and/or request a demo with one of our scientists.
Watch an on-demand webinar showcasing the Crystal and Molecule Optimisers in action.