Molecular Descriptors – key for machine learning in chemistry
Machine learning is a fast growing area of active research within structural science and it is particularly effective in the crystallographic structural sciences due to the wealth of highly accurate structural data available. A key part of machine learning though is having effective molecular descriptors to represent complex chemical information about molecules and structures into easily machine-interpretable vectors of numbers to feed into machine learning algorithms.
 Python scripting
Python scripting
With the recent 2020.0.1 CSD Release launching a series of new molecular descriptors in the CSD Python API, there is now quite a collection of descriptors available for analysing data-sets and for running machine learning projects directly from the CSD Software Portfolio via our Python API. We’re already making use of these descriptors ourselves via a series of research and software development projects towards predicting solid-state behaviour via machine learning.
We already had a range of molecular descriptors available through the CSD Python API in the 2019 CSD Release, including molecular dimensions based on a bounding box (PrincipleAxesAlignedBox), as well as the ability to easily count of numbers of donors, acceptors, rings, rotatable bonds, element types and so on. In the 2020.0.1 CSD Release, we also introduced two families of self returning walk descriptor, a family of topological charge auto-correlation index descriptors, two families for descriptors covering calculated distances between atoms or between element pairs and two families of connectivity index descriptors.
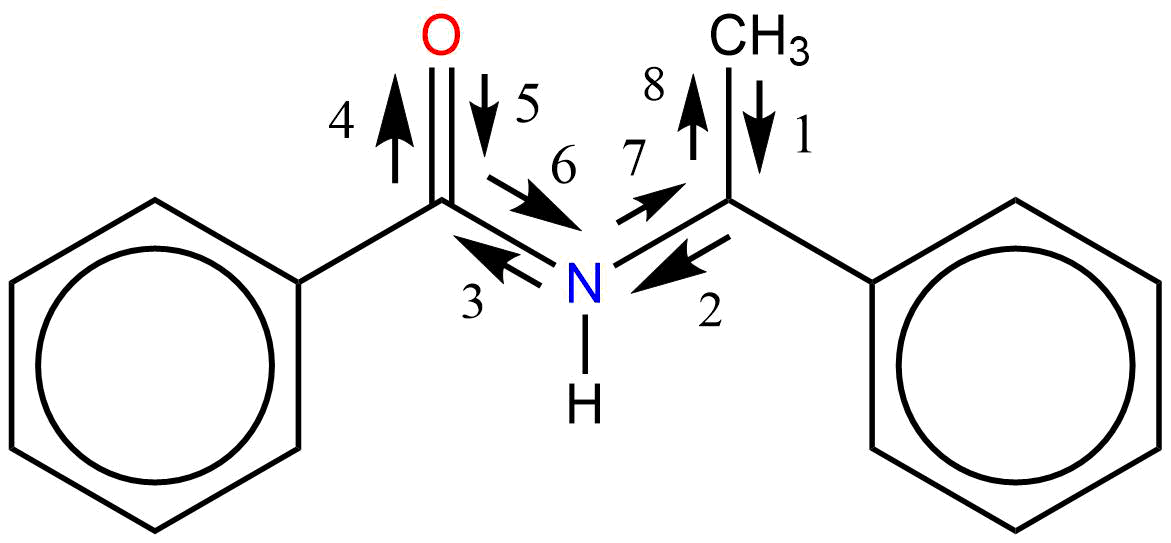
A self-returning walk of length 8 – self_returning_walk(8)
The molecular descriptors offered by CCDC provide representations that can help analyse and understand large volumes of data. For example, the bounding box approach for molecular shape provides the opportunity to define a classification of unit cells based on the ratio of molecular dimensions to cell dimensions. It has been observed that some Box Model patterns are more commonly observed than others – specifically those with a lower surface area to volume ratio. This concept has been used to study the connection between molecular shape and crystal packing[1] as well as isostructurality[2], it has also been employed in creating surface descriptors[3].
Self-returning walk descriptors, connectivity indices and auto-correlation descriptors combined give an excellent description of molecules, aiding in creating descriptive/predictive models, due to their ability in describing molecular size, branching, flexibility and atom/bond types. These descriptors have potential usage not only in descriptive, but also predictive analytics of chemical databases.
All told, this represents quite an array of molecular descriptors that can be produced using the CSD Python API and we expect to continue expanding this descriptor collection in the future to aid both our own machine learning models and those produced by users.
Do let us know what you think about the molecular descriptor functionality available within the CSD Python API, what you’re using the descriptors for and what you’d like to see implemented next. You can email us as always at .
References:
- W. D. S. Motherwell, 2010, 12, 3554-3570. DOI: 1039/C0CE00044B
- L. Fábián, A. Kálmán, Acta Cryst., 1999. B55, 1099-1108. DOI: 1107/S0108768199009325
- , CrystEngComm, 2018, 20, 2698-2704. DOI: 1039/C8CE00454D