Mercury in Action: A Geometry Analysis on Complexes With 2,2′-Dipyridylamine
Here we highlight a work by Samuel Tetteh from the University of Cape Coast, Ghana.
Dr Tetteh investigated the geometry of first-row transition metal complexes with 2,2′-dipyridylamine and halogen ligands by searching the Cambridge Structural Database (CSD) with ConQuest and analysing the data with Mercury. Density Functional Theory (DFT) calculations were also performed to study the stability of the complexes.
The research is part of the Frank H. Allen International Research and Education (FAIRE) Programme, which supports academic researchers and educators from underrepresented communities with access to the wealth of information contained within the CSD.
Why?
The ligand 2,2′-dipyridylamine (dpa, Figure 1 a) is widely used in coordination and organometallic chemistry for its ability to bind to a variety of metal centres and adopt different coordination modes, acting as a monodentate, bidentate, or bridging ligand. Among these, the bidentate coordination mode (Figure 1 b) is particularly stable thermodynamically, and hence it is frequently found in metal complexes.
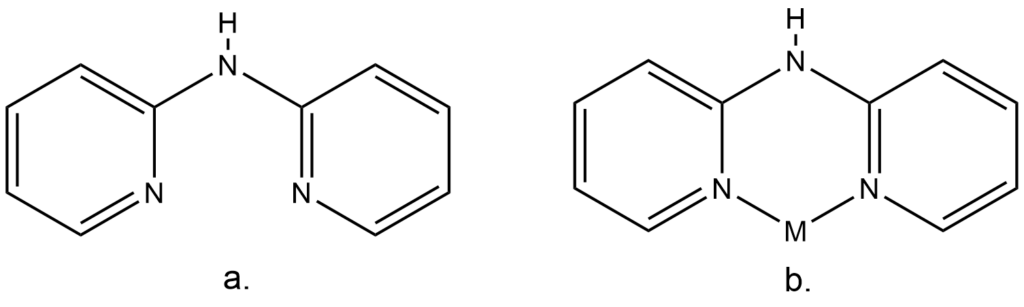
Metal complexes that involve dpa are used as catalysts and other photophysical materials. The ligand is also used for MOFs design and as coligand for magnetic complexes. Despite its popularity, research still needs to be done to understand the chemistry of dpa and its behaviour when in crystal forms.
This work aimed to investigate first-row transition metal complexes with dpa and halogen ligands (F, Cl, Br, and I), and looked at rationalizing their geometry, electronic stability, and reactivity.
How?
Analysis of the Existing Crystal Structures
The study was started by performing a search of the CSD using ConQuest.
Complexes involving any first-row transition metal (1R), with a bidentate dpa ligand, a halogen ligand (7A), and a third ligand of any type (X) were searched. Figure 2 reports the representation of the search query performed. Further filters were applied to only isolate the organometallic structures with determined 3D coordinates.
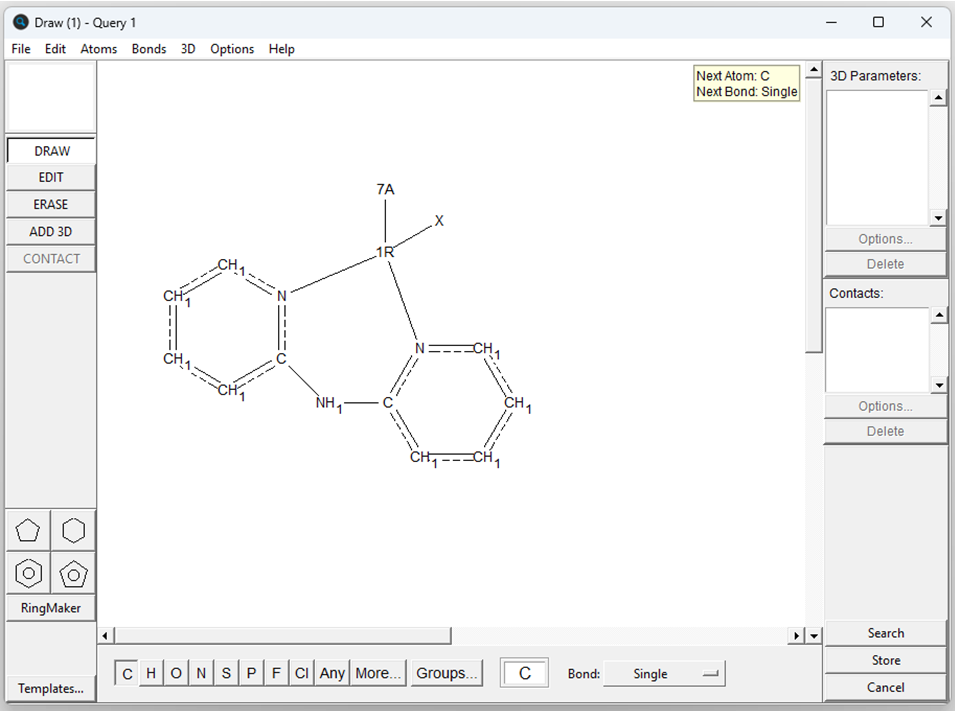
A total of 73 hits were obtained for the query reported above, the majority of which involved copper as metal centre (63 structures). The geometric features of these hits were then explored using Mercury.
An analysis of the distribution of the angles between the pyridyl ring of dpa, the metal centre, and the halogen ligand for these entries led to the identification of two peaks, centred at 95 and 170°. The first corresponded to distorted tetrahedral and square-planar geometries of the complexes; the latter was seen when the pyridyl ring of dpa and the halogen ligand were in trans, with a slight distortion from linearity due to the presence of intramolecular interactions.
The hits obtained from the search of the CSD were also used to investigate the bite angles in the complexes, defined as the angles between the pyridyl rings of dpa and the metal centre (Py‒M‒Py).
It was seen that the values of bite angles for the searched structures went from 77.1 to 94.8°.
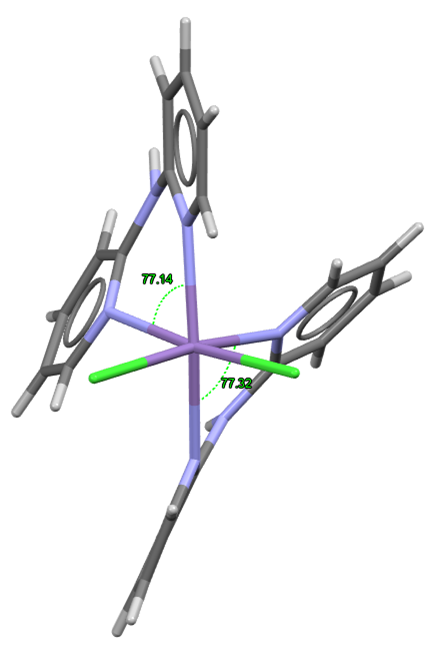
Highlighted in Figure 3 is the Py‒M‒Py angle for CSD Entry SAFCAF, the complex with the smallest bite angle (77.1°). It can be noticed that the deviation from the expected 90° angle, typical for octahedral complexes, is caused by the repulsion of electron density in the pyridyl rings of dpa, demonstrating the flexibility of this ligand.
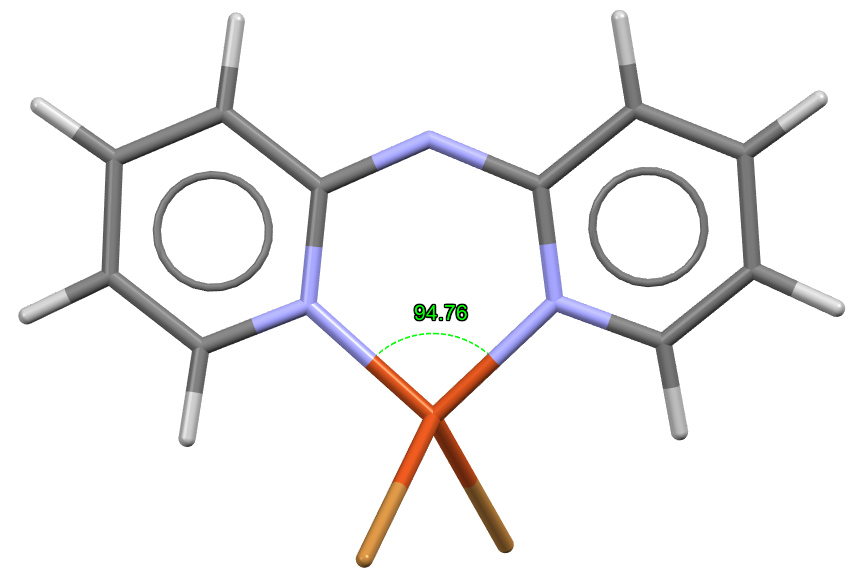
The highest value of bite angle (94.8°) was instead seen for the tetrahedral complex CSD Entry BEHTUD (Figure 4), with a considerable deviation from the expected value of 109.5°. Owing to the nature of bidentate ligand, dpa tends to bind to the metal centre in a cis fashion, restricting the bite angles to lower values.
The average value of bite angle was instead centred around 90°, as expected for bidentate ligands in an octahedral or square planar coordination.
Overall, the variety of bite angles showed that dpa is a flexible ligand that can adapt to different geometries of the metal centre. For more details about other features that were investigated in this work using Mercury, including the intraligand centroid-to-centroid distances in the complexes and the metal-ligand distances, read the full article here.
Analysis of the Stability and Reactivity
Aiming to investigate the stability and reactivity of these compounds, calculations were performed on a series of gas phase tetrahedrally coordinated dpa complexes with halogen ligands.
The calculated metal-ligand bond lengths and bite angles were close to those of the experimental crystal structures in the CSD.
By analysing the gap between the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs), it was identified that the fluoro complexes were the most stable from the halogen series (F > Cl > Br > I). The higher electron density of the iodide ligand is instead responsible for ligand-to-metal charge transfer interactions that lower the HOMO-LUMO gap, making the complex less stable. Important features of the complexes can hence be tuned by choosing different halogen ligands.
Finally, DFT calculations were performed to investigate the chemical reactivities of the metal complexes. Similarly to what seen for the HOMO-LUMO gap, it was shown that complexes with fluoro ligands are less polarizable and hence more stable, while complexes with bulkier halogen ligands exhibited the highest dipole moment and are hence easier to polarize.
Conclusions
This study investigated the geometry of first-row transition metal complexes with dpa and halogen ligands, and examined their stability and reactivity.
The work is part of the CCDC’s FAIRE Programme, which helps support research and education in underrepresented communities.
Next Steps
Read the full article here: Cryst. Growth Des. 2024, 24, 1, 506–513.
Learn more about the Frank H. Allen International Research and Education (FAIRE) Programme.
To discuss further and/or request a demo with one of our scientists, please contact us via this form or .