How can the CSD Ligand Overlay program assist with ligand-based drug design?
How do you design a drug if you don't know the target protein structure? Ligand-based drug design is an approach used in the absence of 3D structures of drug targets. This enables early-stage discovery and lead optimisation even when the protein structure is unknown. One option involves pharmacophore modelling, and it can be used in lieu of a protein-structure-based pathway.
Ligand-based drug design
Pharmacophore modelling is a widely used tool in ligand-based drug design and can provide predictive models suitable for lead compound optimisation. A pharmacophore is a model that captures the nature and three‐dimensional arrangement of chemical functionalities important for molecular interactions with potential biomolecules. (ref) By superimposing a set of active molecules thought to bind to the same target with the same binding mode and then extracting common chemical features that are essential for their bioactivity, researchers can generate pharmacophores. Such a model can then be used to virtually screen libraries of compounds with the aim of identifying potential new binders for the target of interest. Here’s the workflow using CSD-Discovery.
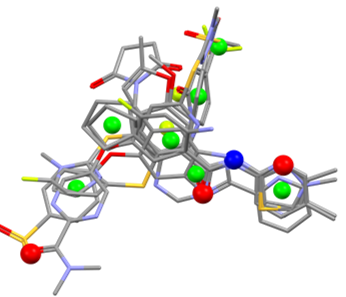
Step one: CSD Conformer Generator
The CSD Conformer Generator can both minimise molecular conformations and generate diverse conformer subsets based on CSD data. The methodology starts from a three-dimensional molecular structure, complete with all hydrogen atoms. The program then samples confirmations based on CSD-derived rotamer distributions and ring templates – returning a diverse set of conformers clustered according to conformed similarity. Each conformer is locally optimised based on features like torsion space and bond angles. The results can serve as inputs for the CSD Ligand Overlay to, for example, generate overlay hypotheses for pharmacophore modelling.
Step two: CSD Ligand Overlay
The CSD Ligand Overlay program superimposes sets of flexible molecules thought to be ligands of the same target biomolecule. It then identifies important molecular features and functional groups, such as hydrogen-bond donors and acceptors, rings and hydrophobic groups. In the absence of any protein-structure information, multiple reasonable overlays for binding might exist. To address this, the program returns multiple diverse overlays that all give potential pharmacophore hypotheses to test. Looking at a few diverse overlay solutions is important – especially in a fully ligand-based approach. Different alternatives can represent plausible pharmacophore hypotheses that can serve as queries during database screening.
Step three: CSD Ligand Screener
The CSD Ligand Screener scans a library of compounds against a pharmacophore query obtained from one or multiple overlaid ligands. The algorithm generalises the 3D pharmacophore definition using atom-property fields created around the query and based on user-defined atom types and potentials. The results are actual molecules with the binding features of the pharmacophore. This can be done programmatically using the CSD Python API.
Case Study: The CSD Ligand Overlay supports the development of new pharmacophores
In this paper, researchers from Universidade Federal do Pará used the CSD Ligand Overlay to develop pharmacophores that may inform new bioinspired competitive inhibitors with pharmaceuticals applications.
Summary
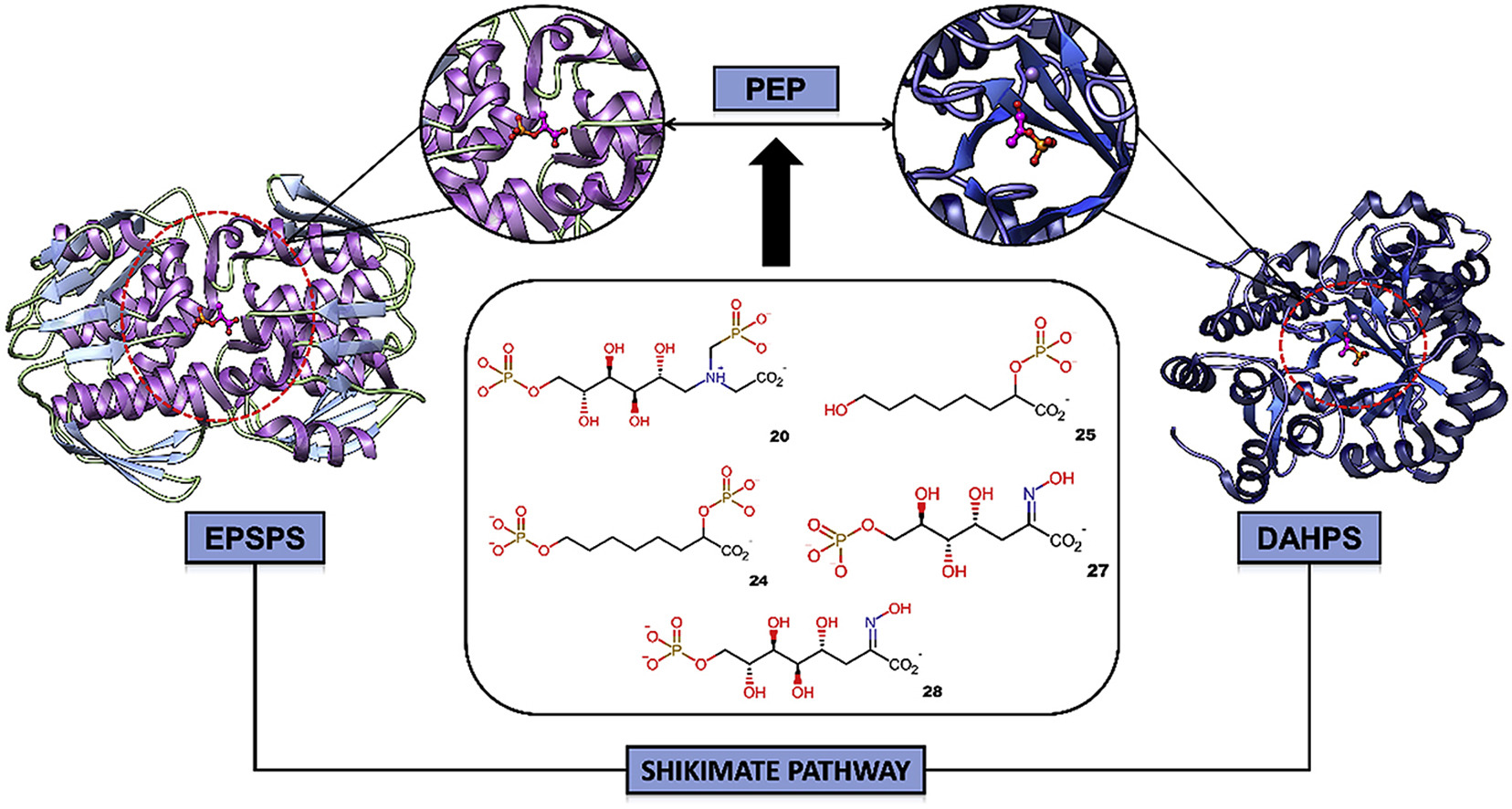
The shikimate pathway is involved with the production of aromatic amino acids, such as phenylalanine, tyrosine and tryptophan. These are essential for plants, bacteria and fungi metabolisms. The 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase (DAHPS) and 5-enolpyruvylshikimate 3-phosphate synthase (EPSPS) catalyze important steps in the shikimate pathway using phosphoenolpyruvate (PEP) as a substrate. Due to the importance of PEP in the shikimate pathway, the team investigated its structure to develop new bioinspired competitive inhibitors against DAHPS and EPSPS that may support the development of antibiotics or antifungals.
How
The researchers performed a literature survey of 28 PEP derivatives, and then analyzed the selectivity and affinity of the compounds against EPSPS and DAHPS using molecular docking, pharmacophore prediction, molecular dynamics (MD) simulations and binding free energy calculations. As part of the process, they used the CSD Ligand Overlay tool to determine pharmacophoric groups of the selected ligands that complex with both EPSPS and DAHPS. Specifically, they generated conformers for each PEP derivative using the Conformer Generator tool and then performed a pharmacophoric prediction using the Ligand Overlay application. The researchers optimized the overlay results based on volume, hydrogen bonding, hydrophobic coplanarity and internal energy scores.
Results
The team found consistent binding modes of the selected ligands and identified pharmacophoric properties related to multi-targets inhibitors for both enzymes. They found a ligand model that exhibited interesting binding energy with EPSPS, one showing an interesting inhibitor forming H-bond interactions with key-residues of PEP and a multi-target inhibitor for both DAHPS and EPSPS.
de Oliveira, M. D. et al. Targeting shikimate pathway: In silico analysis of phosphoenolpyruvate derivatives as inhibitors of EPSP synthase and DAHP synthase. J. Mol. Graph. Model. 101, 107735 (2020).
What’s Next?
Learn more about how the CSD Ligand Overlay enables drug design as part of CSD-Discovery.
Watch this webinar on demand for a demo of the CSD Ligand Overlay as well as helpful tips for getting the most out of the tool.
Want to try it for yourself? Follow the step-by-step tutorial on Ligand Overlay here.