Improved Docking Performance Using Ensemble Docking
This blog aims to illustrate the use of ensemble docking in the context of pose prediction and virtual screening.
Introduction
Over the past two decades there has been an explosion in the number of protein structures available, particularly in the number of protein structures with clinical relevance. This has meant that the key question for people involved in structure based design has changed from “is there a protein model available for my target?” to “how do I best make use of all the structural data available for my target?”. One solution to this problem is to dock into multiple protein models, so called ensemble docking. In this work we will demonstrate how ensemble docking can give significant improvements, both in pose prediction and in virtual screening enrichments.
This example looks at cyclic nucleotide phosphodiesterase (PDE), which cleave phosphodiester bonds in the second messenger molecules cyclic adenosine monophosphate and cyclic guanosine monophosphate. There are many families of PDEs and these tend to have distinct tissue distributions, which make the PDEs attractive drug targets. In this work we will focus on PDE5 which has had several selective inhibitors developed against it including Sildenafil, Tadalafil and Vardenafil. These inhibitors are mainly used as treatments for erectile dysfunction, but are also used in the treatment of pulmonary hypertension.
Method
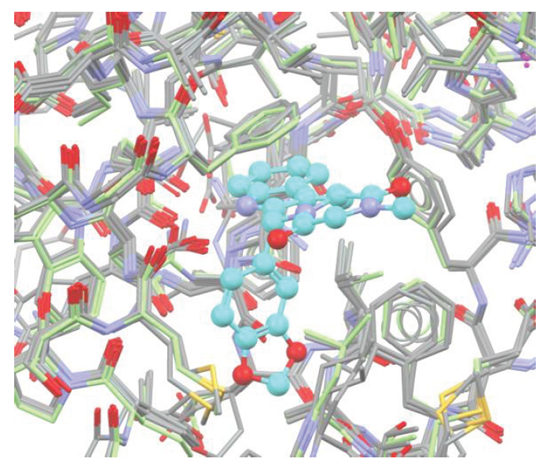
Protein preparation: superimposed and pre-prepared PDE5 structures (1xoz, 1t9s, 1tbf, 1xp0 and 2chm) were obtained from the Astex Non-native Set [1], see Figure 1.
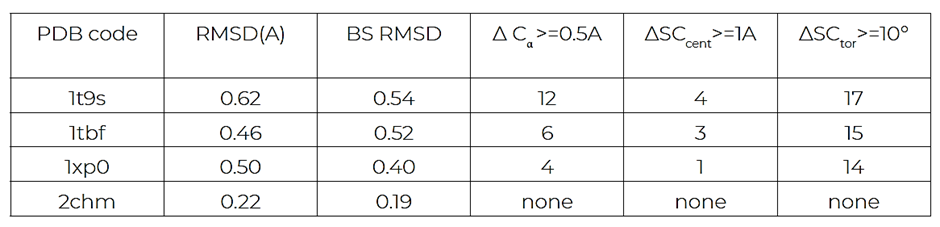
Structural analysis: the structural diversity of the PDE5 binding sites was analysed by using the 1xoz structure as a reference and comparing the root-mean-square deviation (RMSD) of the binding site, the number of Cα backbone movements, the number of side-chain movements and the number of changes in the side-chain torsions. The structural analysis was performed using CSD Cavity comparison tool (accessible through the CSD Python API).
Pose prediction: the 1xoz ligand (Tadalafil) was docked into all the other protein models, i.e. excluding the native 1xoz model.
Virtual screening: the DUD set [2] PDE5 actives and decoys were docked into all the protein models. Area under the curve and enrichment factor statistics were calculated using GoldMine.
Docking setup: the GOLD [3] docking program was used in all docking experiments. The built-in ensemble docking functionality of GOLD [4] was used in both the pose prediction and the virtual screening experiment. The number of genetic algorithm runs was set to 20, early termination was turned off, the solvate all option was turned on and the scoring function was set to ChemPLP. All other options were set to their default values. For purposes of comparison the experiments were also repeated on all protein models individually using traditional single protein docking.
Results
Structural diversity: the protein structures were compared in terms of their RMSDs as well as their Cα and side-chain movements using CSD Cavity API (Table 1). The 2chm protein model was the most similar to that of 1xoz.
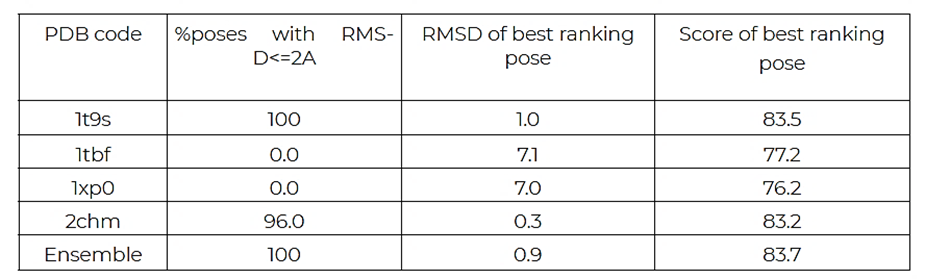
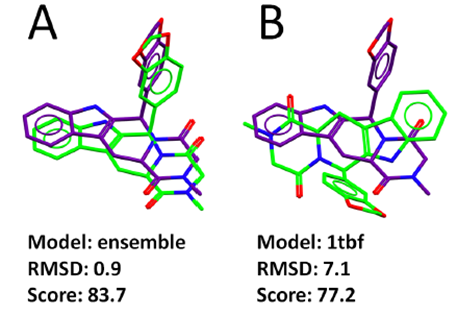
Pose prediction: the results of the pose prediction experiment are presented in Table 2. The ensemble docking run produces the correct pose, the best ranking pose having selected protein model 1t9s. When using single protein model docking the protein models 1t9s and 2chm also produce correct ligand poses. The ligand pose obtained when docking into the 2chm model was in fact even better, in terms of RMSD, than that produced by ensemble docking. However, when using the protein models 1tbf and 1xp0 the correct pose was never found. This highlights the advantage of ensemble docking; it increases the chances of obtaining a correct pose with respect to using a single protein model.
The best ranking poses of Tadalafil produced using ensemble docking and using the protein model 1tbf in a single protein model docking run are shown in Figure 2.
Virtual screening: as illustrated in the pose prediction experiment not all protein models result in correct predictions on a per protein basis. This is also the case for virtual screening experiments where one does not know a priori which protein to dock into in order to get the maximum discrimination performance.
Using the ensemble docking methodology we obtained an area under the curve (AUC) statistic of 0.71 and an enrichment factor (EF) at 5% of 8.5. Figure 3 illustrates how the ensemble docking experiment performed in comparison to docking into the protein models individually using the AUC statistic. Note that the ensemble docking methodology outperforms most of the individual protein models.
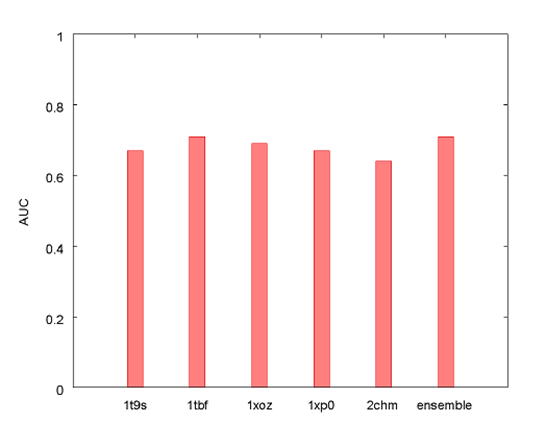
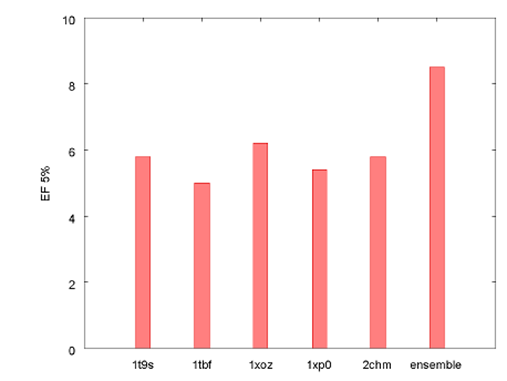
Figure 4 illustrates how the ensemble docking experiment compared to docking into the protein models individually using the EF 5% statistic. In this case the ensemble docking methodology outperforms all of the individual protein models.
Conclusions
In protein-ligand docking it is now common practice to dock into more than one protein model. However, although this tends to give better results, it is not without its complications. The time taken to perform such docking experiments increases linearly with the number of protein models used and so too does the amount of data produced. Furthermore, the complexity of analysing the data increases significantly.
In this study we have shown that the built-in ensemble docking functionality available in GOLD can result in improvements in both pose prediction and virtual screening. Ensemble docking in GOLD makes the selection of the protein model part of the genetic algorithm and can speed up of up to four times compared to sequential docking. It also leads to a reduction in the amount of disc space required. Furthermore, because the selection of the most appropriate protein model is carried out by the genetic algorithm, the data analysis is much simpler with respect to sequential docking, which requires substantial post-processing to identify the best ranking poses from the different docking experiments.
When multiple protein models are available, one does not know a priori which model will give the best docking performance. One strong advantage of ensemble docking is that it very significantly reduces the risk of inadvertently choosing an unsuitable protein model.
In the virtual screening part of this study, the performance of the ensemble docking was actually better than that of the best individual protein model as measured by early enrichment. The fact that ensemble docking can obtain results that are better than its constituent parts stems from the fact that actives that score poorly with respect to decoys in one model, can perform significantly better on a different protein model. Although ensemble docking can provide enrichments better than those of the best individual protein model, this behaviour is not guaranteed.
Next Steps
Find out more about CSD.
Find out more about GOLD.
Find out more about CSD Cavity API.
To discuss further and/or request a demo with one of our scientists, please contact us via this form or .
References
[2] N. Huang, B. K. Shoichet and J. J. Irwin, J. Med. Chem., 2006, 49, 6789-6801.
[3] G. Jones, P. Willett and R. C. Glen, J. Mol. Biol, 1995, 245, 43-53.
[4] O. Korb, S. Bowden, T. Olsson, D. Frenkel, J. Liebeschuetz, J. Cole, J Cheminform., 2010, 2, P25.