Guiding Crystal Habit Modification Using Full Interaction Maps on Surfaces
Investigation of Preferred Interaction Patterns in Differing Crystal Growth Directions Facilitates Strategies for Control of Crystal Morphology.
The particle shape of chemical compounds can have a profound effect on how a powder behaves. Tableting, flow, dispersion, and filtering properties, and even shelf-life depend on the crystal habit or morphology adopted by the crystalline solid state. This is an important parameter to investigate when designing the industrial development process for the production of a drug or medicine.
A crystal habit is determined in the first instance by the arrangement of molecules within the crystal, and different polymorphs or crystal forms can therefore result in particles with different shapes. It is still possible to modify the shape of particles of a given crystal form to improve its manufacturing properties, for example by changing the crystallization solvent or by utilising appropriate additives. The Full Interaction Maps on Surfaces (FIMoS) functionality available in the CSD-Particle suite can help to guide strategies for crystal habit modification.
Full Interaction Maps on Surfaces
The FIMoS capability relies upon the CCDC’s IsoStar software [1]. This is a library of preferred intermolecular interaction geometries, derived for a very broad range of combinations of specific organic functional groups. The library is hence obtained by superposing close interactions from hundreds of thousands of entries in the Cambridge Structural Database (CSD) [2].
FIMoS allows the user to quickly visualize the preferred positions for interactions of a crystal surface with specific organic functional groups of different types. The latter can be hydrogen bond donors and acceptors, or hydrophobic groups. Steric hindrance and surface roughness are also taken into account.
Crystal Morphology Study on Cipamfylline
Cipamfylline (Figure 1) is an anti-inflammatory drug with at least three polymorphic forms. Two of these – forms A and C – exhibit needle-like habits, a shape which can negatively affect critical powder properties like tableting, filtering and powder flow.
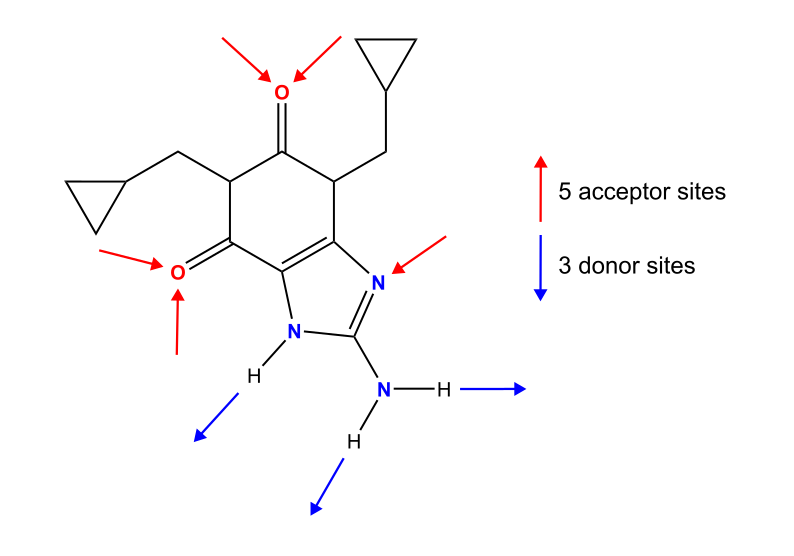
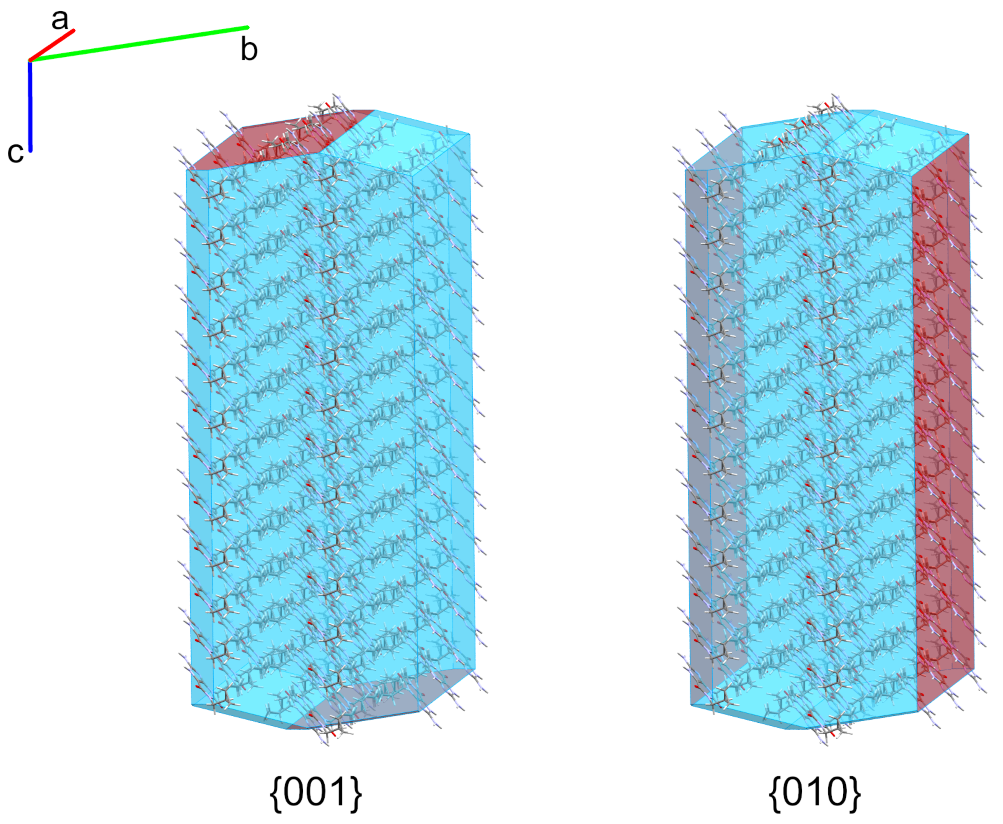
Reported in Figure 2 is the crystal morphology of cipamfylline form A calculated using the Bravais, Friedel Donnay and Harker (BFDH) method [3] in Mercury. The morphology is correctly calculated to be elongated along the c-axis [4], which is indicative of a faster crystal growth along this direction. Slowing the growth of the {001} facets (Figure 2, left), would thus result in less elongated crystal particles with improved manufacturing properties.
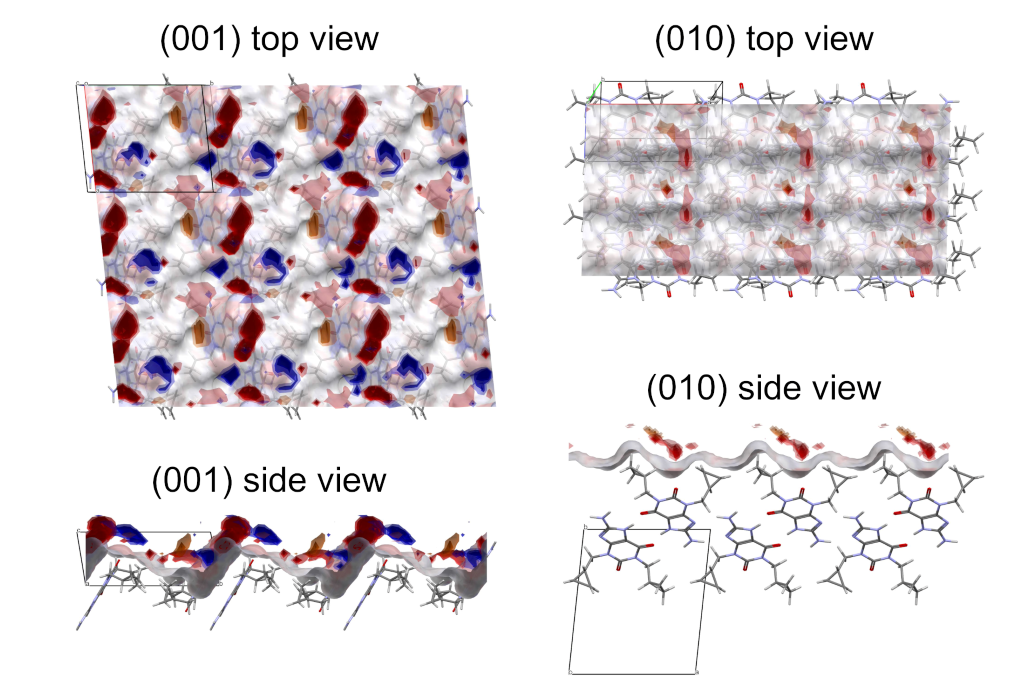
To further investigate the surface properties of cipamfylline, the FIMoS functionality was used. Figure 3 shows a comparison of FIMoS calculated for the (001) and (010) surfaces. The (001) surfaces display a high density of hydrogen bond donor and acceptor groups, as well as availability to engage in aromatic interactions. The (010) surfaces instead, which grow in the perpendicular direction, have considerably less sites for interacting with incoming molecules. This suggests a strategy to design a growth inhibitor for cipamfylline. For example, an additive that interacts strongly with the (001) surfaces, but not with the (010) surfaces, would result in crystal particles with improved properties.
Conclusion
Full Interaction Maps on Surfaces calculated with Mercury can assist in designing strategies to modify particle shapes.
Next Steps
For further information about Full Interaction Maps, see Wood et al. [5].
Follow the link to find out more about how to visualize the BFDH Morphology in Mercury.
To discuss further and/or request a demo with one of our scientists, please contact us via this form or .