A Million Crystal Structures: the CSD Contribution in the Designing of Biologically-Active Molecules
Recently our colleague Pete Wood, in collaboration with Robin Taylor, authored an exhaustive review on how the Cambridge Structural Database (CSD) has proved to be an invaluable resource for the design and development of new drugs, as well as for the design of commercially relevant materials such as dyes and energetic materials. You can read the full review ‘A Million Crystal Structures: The Whole is Greater than the Sum of Its Parts’ here.
It was a very timely review as its publication almost concurred with the time when the millionth crystal structure was added to the CSD. What a perfect moment to recollect and share with the scientific community the value of this highly curated database and the insights that can be gained from it.
Among all the great research reviewed in the article, I was particularly intrigued by how, in the last decade, the CSD was used to aid in the design of biologically-active molecules.
Here, I will highlight few successful applications of the CSD in the field.
Drug designers started to make use of the CSD back in the eighties, with the aim to learn more about molecular geometries and interactions. Later, the CSD also became a provider of data needed for the development of novel drug-design software such as LUDI [1] and DOCK [2] .
Nowadays, both drug designers and client applications still heavily use the CSD. It remains an invaluable resource when it comes to understanding molecular shapes and molecular recognition, and it’s undoubtedly a diverse 3D chemical database.
Understanding molecular shapes
Protein-ligand recognition is driven by two key aspects: shape complementarity and molecular interactions. Being able to predict the energetically accessible molecular geometries is, therefore, of crucial importance in rational drug design. Unlike force fields and quantum mechanical calculations, knowledge-based methods derived from crystallographic data have the advantage of being based on precise, experimental information. Furthermore, molecular conformations in a condensed phase, like those in the CSD, tend to resemble extended conformations adopted by ligands when bound to a protein. Small-molecule crystals are held together by favourable intermolecular interactions (e.g. hydrogen bonds) rather than intramolecular forces. In contrast, theoretical in vacuo energy calculations are biased toward folded conformations, which enable favourable intramolecular contacts to be made.
The vast literature produced by scientists over the years about the value of the CSD on assessing conformational preferences, concluded that torsion information derived from small-molecule crystal structures can provide useful hints for designing molecules with desired shapes. Applications of the CSD span from the selection of linking groups, to docking-solution validation, to conformational locking via intramolecular H-bonding, to rationalisation of inactivity.
An illustrative example of how advanced searches in the CSD can assist to better understand the conformational spaces was published by a team from Roche Basel. Knowledge derived from the CSD enabled rationalisation of the inactivity of benzamide inhibitors of tissue factor/factor VII (TF/F.VIIa). With the aim of generating selective inhibitors of TF/F.VIIa, rational drug design resulted in compounds 1 and compound 2 of Figure 1. Design was meant to optimise the well-known binding mode of ligands to this target which sees the benzamidine moiety binding to the S1 pocket and the phenyl ring in the S3 pocket, with the addition of a small alkyl group to the benzylic position, in order to occupy the S2 pocket that is specific for the F.VIIa active site.
However, neither one of the two designed inhibitors showed any activity against TF/F.VIIa. Detailed investigations of the anticipated binding mode showed that the required nearly coplanar arrangement between the alkyl group and the amide nitrogen in the C(sp2 )−C(CH3)− CO−NH− fragment, was not favourable.
CSD searches for the corresponding fragment showed that the particular coplanar geometry was very unlikely, and the fragment would rather have a strong preference to adopt torsion angles in the range of 120-150° (see top histogram in Figure 1). In contrast, the related mandelic acid fragment C(sp2 )−C(O)− CO−NH−, where the alkyl group is replaced by an alkoxy group, showed a preference for the desired rotamer (bottom plot in Figure 1). This finding led to the design of a new 11 μM inhibitor (compound 4 in Figure 1), giving entry to a new class of inhibitors that have the desired conformational preference to selectively bind F.VIIa (Brameld etal., J. Chem. Inf. Model. 2008, 48, 1−24. DOI: 10.1021/ci7002494).
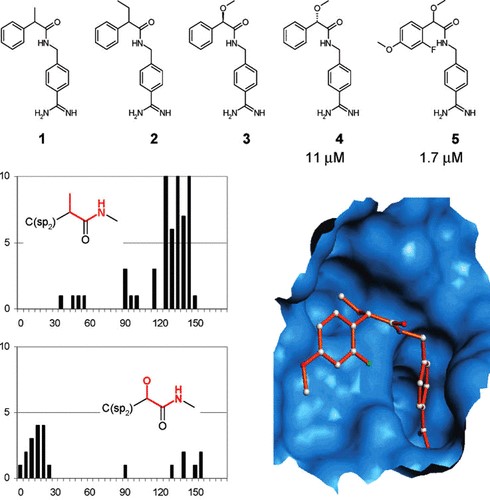
Figure 1: Rational design of TF/F.VIIa inhibitors. Compounds 1 and 2 were found to be inactive as inhibitors because their preferred conformations do not fit the active site. The mandelic acid analogues (4 and 5) have different conformational preferences that allow the molecule to bind the TF/F.VIIa.
Understanding Molecular Recognition
The strong publication record demonstrating CSD applications to rationalise molecular geometries is highly complimented by examples where the fully curated crystallographic information derived from the CSD has been used to analyse molecular interactions.
Understanding and optimising the interactions between a ligand and the protein binding site is fundamental in many research applications including rational drug design and crystal engineering.
However, as Wood and Taylor state in the review, despite the undiscussed value of the CSD, two issues should be considered when non-bonded contacts in small molecule structures are used to predict what might occur between proteins and ligands. First, contacts across crystallographic inversion centres are often favoured by crystal packing and may not always be good models for interaction likelihoods in chiral environments. Second, the ratio of hydrogen bond donors to acceptors is much higher in proteins than in small-molecule crystal structures, so H-bonds to weak acceptors are more likely to form in protein−ligand complexes than in CSD structures.
IsoStar, the intermolecular contacts knowledge base library derived from CSD and PDB data, stores scatterplots that reveal information about how functional groups interact with each other. Such scatterplots are the results of searches in the CSD, or PDB, for non-bonded interactions between a pair of functional groups. Figure 2 is an example of IsoStar scatterplots, it shows the distribution of OH and NH groups around 1,3,4-thiadiazole ring. Two conclusions can be drawn from that plot: (a) the nitrogen atoms are good but directional H-bond acceptors—it is rare for donor groups to point to the middle of the N=N bond—and (b) the thiadiazole sulfur rarely, if ever, acts as an acceptor.
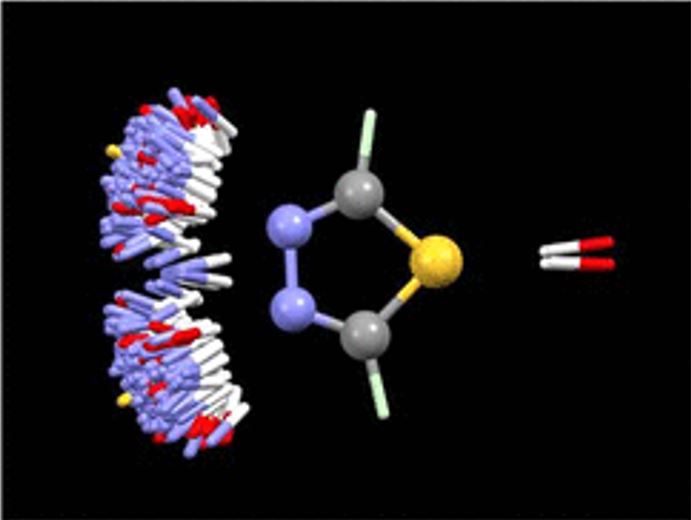
Figure 2: CSD-based IsoStar plot of N–H and O–H distribution around 1,3,4-thiadiazole. Each contact is shown twice, the extra one being generated by reflection in the mirror plane of the thiadiazole moiety perpendicular to the plane of the figure.
IsoStar information on the frequencies and directionalities of intermolecular contacts is particularly relevant to medicinal chemists interested in identifying bioisosteric replacements, and to molecular modellers engaged in structure-based drug design. To fulfil these requirements some programs have been developed on top of the IsoStar library (i.e. Full Interaction Maps (FIMs), SuperStar and Fragment Hotspot Maps). They help scientists to gain insights about intermolecular interaction patterns between functional groups in the context of either small-molecule crystal structures (FIMs) and protein-ligand complexes (SuperStar and Fragment Hotspot Maps).
SuperStar has been particularly useful to identify potential binding hotspots in proteins and ligands. It uses crystallographic information to generate composite propensity maps for protein binding sites by estimating the probability of an interaction between a protein and a small functional group (such as methyl or carbonyl group), based on how often the interaction has been observed in the IsoStar library. SuperStar has been extensively used in drug discovery to validate hypothesized docking poses and to identify structural waters in protein binding sites.
However, SuperStar indicates where single interactions can occur, not where whole fragments can be placed. To overcome this limitation, a modified algorithm has been developed, where three SuperStar maps are simultaneously generated to indicate favourable grid points for carbonyl oxygen, uncharged NH, and aromatic CH. Deeply buried grid points, corresponding to regions of high SuperStar scores, are then examined to produce fragment hotspot maps. These maps highlight pockets that are sufficiently big to accommodate the probe atom (e.g. carbonyl oxygen) and an attached hydrophobic fragment in a preferred orientation for binding. Figure 3 shows the stages in the calculation of the fragment hotspot maps.
An application of this program was recently published by Roca et al. from the Biological Research Center in Spain (Roca et.al, J Enzyme Inhib Med Chem, 2018, 33, 1034-1047 DOI: 10.1080/14756366.2018.1476502.) to identify allosteric sites of acetylcholinesterase. Since allosterism represents a powerful means to regulate non-classical protein functions, allosteric sites of acetylcholinesterase were targeted to design more selective molecules that will modulate the non-hydrolytic functions of cholinergic system.
In this study, they used the fragment hotspot maps programme to identify the key residues that would allow a fragment to effectively bind to two predicted allosteric cavities of the acetylcholinesterase. The combination of the fragment hot spot maps programme with other computational and experimental tools led to the identification of several allosteric compounds able to modulate the non-hydrolytic functions of the acethylcolinesterase.
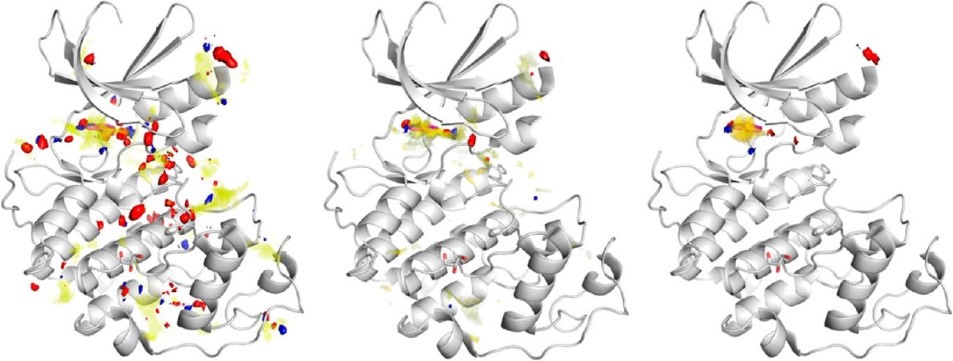
Figure 3: Example enzyme with atomic SuperStar propensity (left), propensities weighted by degree of burial (center), and fragment hotspot map (right). Yellow, hydrophobic map; blue, H-bond donor; red, H-bond acceptor.
Optimising Aqueous Solubility
In recent years, CSD interaction data found a promising application during the lead optimisation phase of the drug discovery process. Optimising solubility is a continuous challenge for the pharma industries. It has been reported that more than 40% of new chemical compounds developed in the pharmaceutical industry are practically insoluble in water.
Two key determinants of low aqueous solubility are: (i) poor solvation (i.e., unfavorable solute-solvent interactions in solution) and (ii) high crystal lattice energy (i.e., strong solute-solute interactions in the solid state). CSD interaction data can be used to suggest ways of improving the latter as notably shown by Scott et al. from AstraZeneca (Scott et al., J. Med. Chem. 2012, 55, 5361–5379. DOI: 10.1021/jm300310c).
They inspected the molecular interaction pattern in the crystal structure of a potent, but poorly water soluble GPR119 agonist and observed a peculiar head-to-head intermolecular interaction between aryl-bound methylsulfone groups (Figure 4). A search in the CSD proved that this was a very common motif and hinted that such a packing arrangement could contribute to the strong lattice energy of the compound and therefore the undesirable solubility. Indeed, replacing the aryl methyl sulfone moiety with a cyanopyridyl group retained the potency and increased the solubility by 2 orders of magnitude.
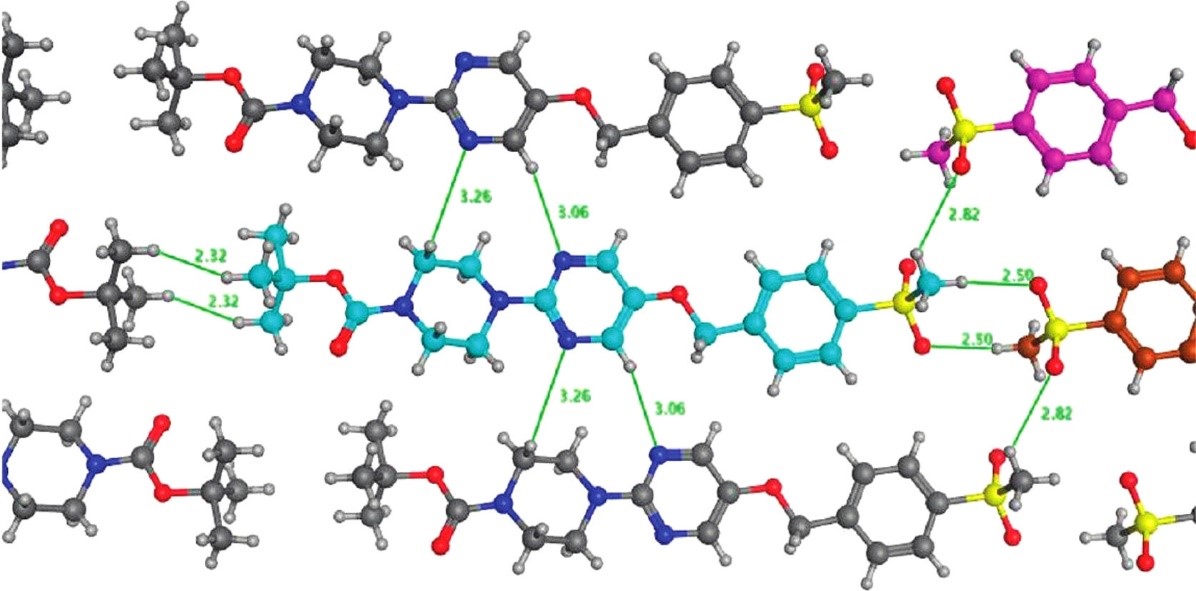
Figure 4: Motifs in the crystal structure of a potent GPR119 agonist. The head-to-head motif between methylsulfone groups occurs very frequently in the CSD and was deduced to be a key factor in increasing lattice energy thereby reducing solubility.
The CSD as a Diverse Chemical Database
Telling how diverse (or similar) structures are within a chemical database is very subjective. The rationale behind Wood & Taylor’s description of the CSD as a diverse database is the fact that if a synthetic chemist makes an interesting or unusual compound, you can be sure that he or she will ask for its crystal structure to be solved and this structure will be then included in the CSD.
Thus, the CSD is replete with novel molecules and it is an attractive database to use for searches designed to find new leads for drug discovery projects. In addition, 3D searches of the CSD will have the advantage that the molecular conformations therein are proven to exist. However, on the flip side, hits resulting from CSD searches might be interesting but physical samples are unlikely to be available for testing. Therefore, the diversity of the CSD can be fully utilized in projects where synthetic effort is available.
Different types of database search techniques exist (e.g. pharmacophore searching, scaffold hopping, searches for bioisosteres or linker groups, shape matching) with some obvious overlap between them which makes it difficult to precisely assign a given search to the appropriate class.
An interesting example of scaffold hopping after using the fragment libraries derived from the CSD was described by Kuhn et al. (Kuhn et al., J. Med. Chem. 2016, 59, 4087– 4102, DOI: 10.1021/acs.jmedchem.5b01875). In this work the researchers wanted to replace a meta-substituted phenyl ring in the centre of a known BACE1 inhibitor (compound 19 in Figure 5) with a more polar linking group in order to improve the physicochemical properties. Fragments were searched in the CSD for the best possible replacement – whilst keeping all connected residues, i.e., the rest of the query compound in place, using the ReCore program distributed by BioSolveIt.
ReCore search suggested a trans-cyclopropylketone linker as a polar replacement to a meta-substituted phenyl ring (CSD entry FUQGAZ), that looked an almost perfect geometric match. Incorporating this fragment into the original compound led to a slightly more potent BACE1 inhibitor with lower log D and improved aqueous solubility (compound 20 in the Figure 5).
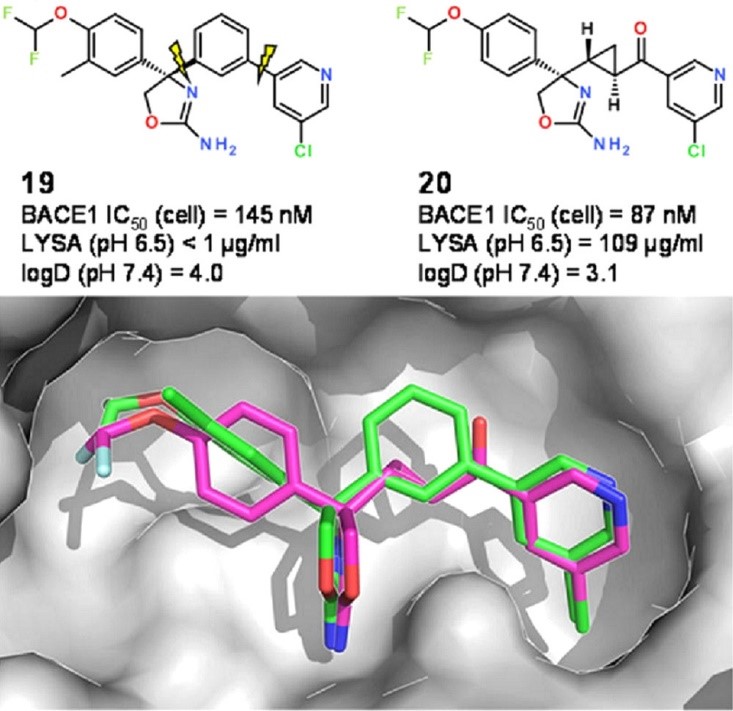
Figure 5: Original BACE1 inhibitor (19) and the planar analogue (20) containing a replacement fragment suggested by CSD entry. Adapted from (J. Med. Chem. 2016, 59, 4087– 4102, DOI: 10.1021/acs.jmedchem.5b01875). Copyright 2016 American Chemical Society.
Conclusions
This blog is a short walk through a few of the many examples showcasing successful uses of the CSD in several areas, including drug discovery and development. The chemical coverage of compounds in the CSD increases year-on-year as new classes of compounds are synthesized and crystallised. Consequently, the detailed insights from data have become discernible. We at the CCDC, together with the scientific community, are excited about the new challenges that may come, and look forward to the next million structures and the insights they will provide.
Images source: Chemical Reviews, A Million Crystal Structures: The Whole Is Greater than the Sum of Its Parts
[1] ) Böhm, H.-J. The Computer Program LUDI: A New Method for the de Novo Design of Enzyme Inhibitors. J. Comput.-Aided Mol. Des. 1992, 6, 61−78
[2] Kuntz, I. D. Structure-Based Strategies for Drug Design and Discovery. Science 1992, 257, 1078−1082.