CSD in Action: How Metal Ammine Complexes Interact With Aromatic Rings
Here we highlight a paper by Snežana D. Zarić and co-workers from the University of Belgrade.
In this work, the team analysed the Cambridge Structural Database (CSD) and used quantum chemical calculations to perform an in-depth study of the NH/π interactions between coordinated ammonia (NH3) and C6-aromatic rings.
Why?
NH/π interactions are common in nature. They are found in proteins and related structures, and are involved in important mechanisms such as the transport of ammonia through the cell membrane.
While the interactions of non-coordinated ammonia with aromatic rings are well studied, the NH/π interactions found between metal ammine complexes (metal complexes containing at least one ammonia ligand) and C6-aromatic rings have not been thoroughly explored yet.
This work provides a systematic study of the factors that influence the NH/π interactions from coordinated ammine ligands with C6-aromatic rings, which includes:
- the number of coordinated ligands that interact with the aromatic ring;
- the metal complex charge;
- the size of the metal;
- the coordination number of the complex.
How?
Searching the Data in the CSD
The team started by searching the CSD for NH/π contacts involving C6-aromatic rings and both non-coordinated ammonia and metal ammine complexes. The search was performed using ConQuest, and led to the identification of:
- 48 contacts involving a non-coordinated ammonia interacting with a C6-aromatic ring (Group A);
- 354 contacts involving one coordinated ammine ligand interacting with a C6-aromatic ring (Group B);
- 56 contacts involving two ammine ligands (coordinated to the same metal centre) interacting with a C6-aromatic ring (Group C);
- 8 contacts involving three ammine ligands (coordinated to the same metal centre) interacting with a C6-aromatic ring.
Given the small sample, the latter were not used in the rest of the study. For further details on the criteria used to perform the search, refer to the full article here.
The team then started to investigate the structures obtained from the search, aiming to understand how the charge of the interacting species impacts the strength of the NH/π interactions.
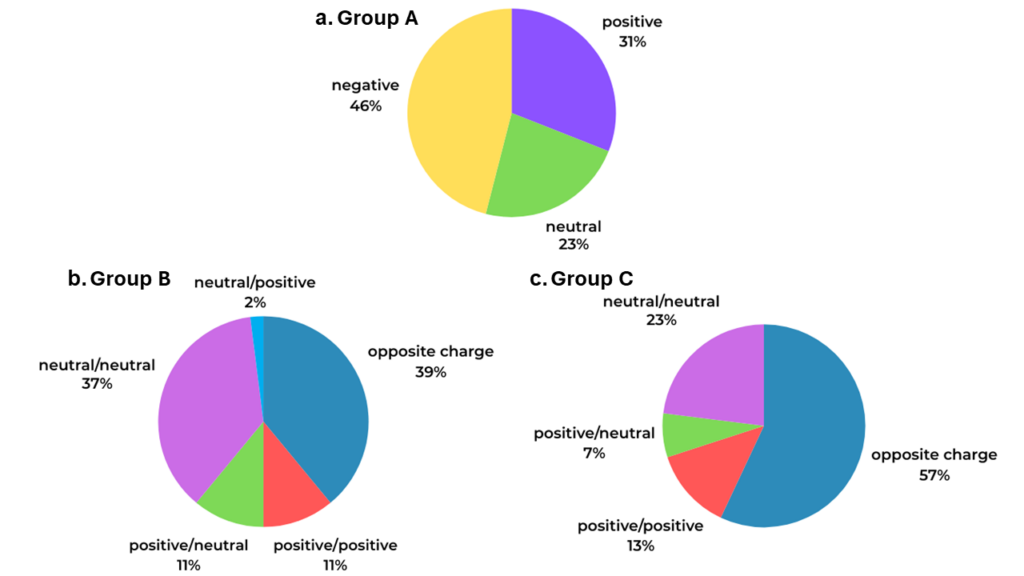
While the non-coordinated ammonia in Group A is always neutral, the C6-aromatic ring can be part of a neutral, positive, or negative species. From Figure 1 a. it can be seen that almost half of the crystal structures included in Group A involve an aromatic ring that is negatively charged (46%), supporting the observation that these are the best hydrogen bond acceptors in NH/π interactions.
In Group B and C, instead (Figure 1 b. and c.), both the interacting species can be either neutral or charged.
From Figure 1 b. it can be seen that most of the crystal structures included in Group B involve a coordinated ammine ligand and a C6-aromatic ring with opposite charge (39%), or neutral species (37%). Additionally, while a positively charged ammine complex strengthens the NH/π interactions (Figure 1 b. green and red), a positively charged C6-aromatic ring weakens them (Figure 1 b. light blue).
For NH/π interactions involving two coordinated ammine ligands and a C6-aromatic ring (Group C, Figure 1 c.), in more than half of the contacts seen in the CSD the interacting species have opposite charge (57%), while only 23% of the contacts involve neutral species.
The team then looked at the geometric parameters of the crystal structures belonging to Group B (the most populated group), where hence one coordinated ammine ligand and a C6-aromatic ring interact. In particular, they looked at the distributions of the distances between the interacting hydrogen atom from the ammine ligand and the aromatic ring centre. This is an overview of the results obtained:
- Structures with interacting species with opposite charge showed a peak between 3.0 and 3.5 Å, but are the dominant contacts at short distances (2.0‒3.0 Å) and are hence the strongest interactions;
- Structures with neutral interacting species showed a peak between 3.0 and 3.5 Å, but are less dominant than the ones with opposite charge at short distances;
- Structures with a positively charged ammine ligand and a neutral aromatic ring showed a broader peak (2.5‒3.5 Å) from rather short distances, and can hence be considered stronger than the ones involving neutral interacting species;
- Structures with positively charged interacting species showed a peak at longer distances, between 4.0 and 4.5 Å, and are hence the weakest.
The Quantum Chemical Calculations
Aiming to identify the most stable interaction geometries, the team calculated the potential energy surfaces for the cadmium complex [Cd(NH3)6]2+, which is present in the CSD for interactions involving one, two and three ammine ligands, and a benzene ring. It was shown that systems with a horizontal displacement of 0.0 Å (where the hydrogen atom of the ligand, or the centroid between the two or three hydrogen atoms of the different ligands, is directly above the aromatic ring centre) were the ones with minima of potential energy. At the minima, the strongest interactions are seen in systems involving three interacting ligands, while the weakest are seen in those involving one interacting ligand. More details can be found in the full article.
Quantum chemical calculations were then performed to study the NH/π interactions on models involving ammine complexes with different coordination geometries (octahedral, tetrahedral, square-planar, and linear) and the benzene ring. Different metals were hence involved, having different sizes; a variety of coordination numbers for the complexes were investigated; a diverse number of interacting ligands and complexes with different charge were also considered.
The team calculated the interaction energies on these models using a horizontal displacement of 0.0 Å (calculated to be the minima, as shown above). This is an overview of the results obtained:
- In agreement with what seen from the analysis of the structures in the CSD on the influence of charged metal complexes on the strength of NH/π interactions, the higher the metal complex charge, the stronger the interaction;
- Considering the same metal complex, when three ligands interact with a C6-aromatic ring the interaction is stronger, followed by the system with two interacting ligands, and finally the system with one interacting ligand;
- When complex charge, coordination numbers, and number of interacting ligands are the same, choosing a smaller metal centre strengthens the NH/π interactions, as less steric clashes between the complex and the benzene ring are present;
- When complex charge and metal oxidation number are the same, choosing a complex with a smaller coordination number strengthens the NH/π interactions, as the positive metal charge is partially dispersed to a smaller number of ligands;
- Overall, NH/π interactions involving coordinated ammonia and benzene are notably stronger than those involving non-coordinated ammonia and benzene.
Conclusions
An in-depth search of the crystal structures in the CSD allowed the scientists to study the NH/π contacts involving metal ammine complexes and C6-aromatic rings, and discover important features of these interactions.
By combining the search of the database with quantum chemical calculations, the team could not only identify which are the strongest and weakest NH/π interactions, but also discover how factors such as the complex charge or coordination number influence their strength.
Next Steps
Read the full article here: Cryst. Growth Des. 2024, 24, 4, 1705–1714.
To discuss further and/or request a demo with one of our scientists, please contact us via this form or .