CSD in Action: Discovery of a Potent Selective Estrogen Receptor Degrader Antagonist for the Fight Against Breast Cancer
With around 2 million new cases and 600,000 deaths worldwide per year, breast cancer is the most commonly diagnosed type of cancer among women [1].
This blog presents the work carried out by researchers from AstraZeneca, who used the Cambridge Structural Database (CSD) to help in identifying a highly potent selective estrogen receptor degrader antagonist for the treatment of breast cancer.
Introduction
Estrogen receptor α (ERα) is a transcription factor that is involved in the growth of breast epithelial tissue. ERα has a central role in hormone-receptor-positive (HR+) disease, which is present in 70% of breast cancer patients. Numerous endocrine therapies to target ERα have therefore been explored.
One of the classes of drugs that proved efficient in targeting ERα is called selective estrogen receptor degraders (SERDs) antagonists, which includes Fulvestrant as the only approved drug. Fulvestrant is an ERα antagonist that causes the degradation of the ERα protein and has been shown to be effective in HR+ patients. However, its low oral bioavailability represents a major limitation, and this has led to rising interest in the discovery of new orally bioavailable SERDs.
This work reports the optimization of a meta-substituted series of SERD antagonists, and the identification of a highly potent compound with good oral bioavailability and favourable physicochemical properties.
Discussion
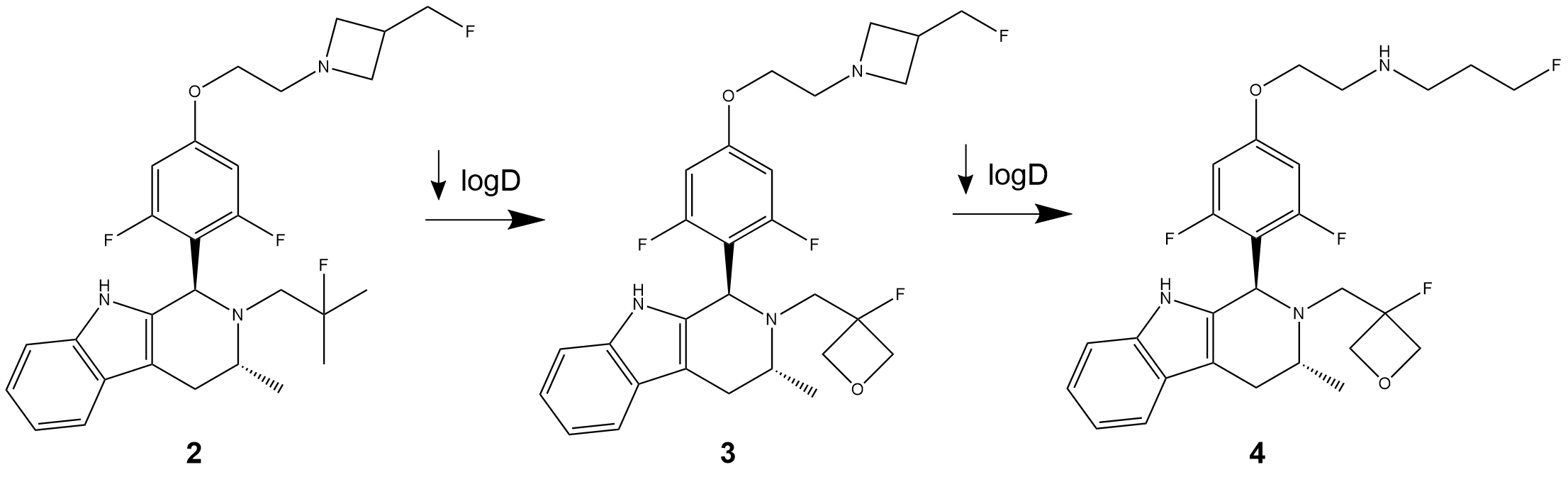
Reported in one of their previous works [2], the scientists from AstraZeneca identified compound 2 (Figure 1) as an optimal starting point to perform a structural optimization that can lead to a chemically distinct series of new oral SERDs. In fact, compound 2 presented an interesting pharmacological profile, but its physicochemical properties were not optimal due to its high lipophilicity (log D7.4 >4.4).
Structure-activity relationship (SAR) analysis allowed the researchers to obtain compound 3, where the introduction of the oxetane ring led to a lower lipophilicity while maintaining high drug potency. Further optimization was seen by introducing an acyclic amine to form compound 4. This led to an additional decrease of lipophilicity, while once again maintaining the potency and degradation values of the initial compound.
The group noticed that most of the SERDs reported in the literature that progressed to the clinic stage, presented substituents in the para position of the pendant phenyl ring. Even if the mechanism of degradation of the ERα protein is unknown, it was hypothesised that the binding of a SERD would be followed by a structural transition form of the protein, which would be flagged for degradation.
Aiming to search for alternative moieties capable of initiating the degradation sequence, the scientists inspected the structures of Erα available in the literature and investigated the interactions at the substituent in para position.
CSD in Action
An approach that differs from the SAR analysis was also used. From a fragment of a SERD that reached phase III clinical trials containing a para-base binding pose, the scientists constructed an energy-minimized ring-based pharmacophore. This was then used to perform a search for small-molecule crystal structures present in the CSD.
The approach allowed them to search for motifs that differ from the one present in the starting fragment, but that reliably denote a conformation that is close to the minimum of energy. From two examples of compounds obtained from the search it was identified that basic groups on a short chain, and substituent in the meta position, are preferred. These pharmacophore hits mimic the orientation of the original para-linked base but are substituted in the meta position.
Modeling of the meta-linked basic group hits allowed the scientists to confirm that they fit in the pocket, and to identify the meta-base orientation that facilitates interactions over the other possible directions.
The group proceeded by then modifying the scaffold and further optimizing the fragment structure to ensure that the investigated drugs would exhibit a good pharmacological profile. Putting together the observations made with the SAR analysis, the pharmacophore search, and the NMR conformational analysis, researchers selected compound 38 (see article here: J. Med. Chem. 2023, 66, 4, 2918–2945) as a promising candidate.
Compound 38 forms a short contact between the carboxylate of the side chain and His524, and it is in close proximity to Asp351. This compound was revealed to be a highly potent SERD, which showed excellent capability to degrade Erα and a good oral bioavailability.
Next Steps
Find the full article here: J. Med. Chem. 2023, 66, 4, 2918–2945.
To discuss further and/or request a demo with one of our scientists, please contact us via this form or .
References
[1] Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R.L., Torre, L.A. and Jemal, A. (2018), Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 68: 394-424.
[2] Scott, J. S.; Breed, J.; Carbajo, R. J.; Davey, P. R.; Greenwood, R.; Huynh, H. K.; Klinowska, T.; Morrow, C. J.; Moss, T. A.; Polanski, R.; Nissink, J. W. M.; Varnes, J.; Yang, B. Building bridges in a series of estrogen receptor degraders: an application of metathesis in medicinal chemistry. ACS Med. Chem. Lett. 2019, 10, 1492−1497.