CCDC Research on COVID-19
The CCDC is actively seeking areas where we can assist in the fight against the nefarious SARS-Cov2 disease currently taking hold around the world.
We’ve already started committing to research time and have noted several collaborative efforts.
How can the CCDC help?
Our team, software and network make us well positioned to support research efforts into COVID-19 treatments.
The scientists at the CCDC span many specialities within drug discovery and development, and have many years of experience from both pharmaceutical and academic sides. Early on in the pandemic we monitored updates and began thinking of ways we could contribute.
When combined with our software (encompassing various tools to identify, refine and test drugs in silico) and our network of collaborators and users we hope to contribute to this global fight – and we’ve already begun.
.
Refining publicly available information
As people with an interest in data, our first undertaking has been to try to understand the structural information that is being generated worldwide.
In order to have the best chances of success in finding new therapies, two recent protein structures have been published of the COVID-19 main protease with inhibitors bound (PDB entries 6LU7 and 6Y7M). Necessarily, the researchers in this space have pushed these structures into the public domain quickly to help others.
That said, we noted when we looked at these structures that the bound ligand geometries had some features that the CCDC tool Mogul would deem somewhat unusual, so we asked our collaborators at Global Phasing to take a look. The team there have started to re-refine 6Y7M using their in-house workflow. They are trying to improve the underlying fit of the structures to the data and the quality of the compound geometries as judged by Mogul.
We hope these re-interpretations of the data will be useful to the community; you can see the on-going work on their wiki here.
.
Understanding the binding site
CCDC has a project to develop a method for informatically analysing binding sites (See our recent publication https://pubs.acs.org/doi/10.1021/acs.jcim.9b00996).
Our sponsored PhD students, Mihaela Smilova (Oxford) and Peter Curran (Cambridge and UCB) have been looking at using these methods to characterise the pharmacophore from the structural data. They’ve generated a visualisation of the pocket contoured at various levels, identifying key binding points for inhibitors based on the set of structures generated by the XChem team at Diamond.
.
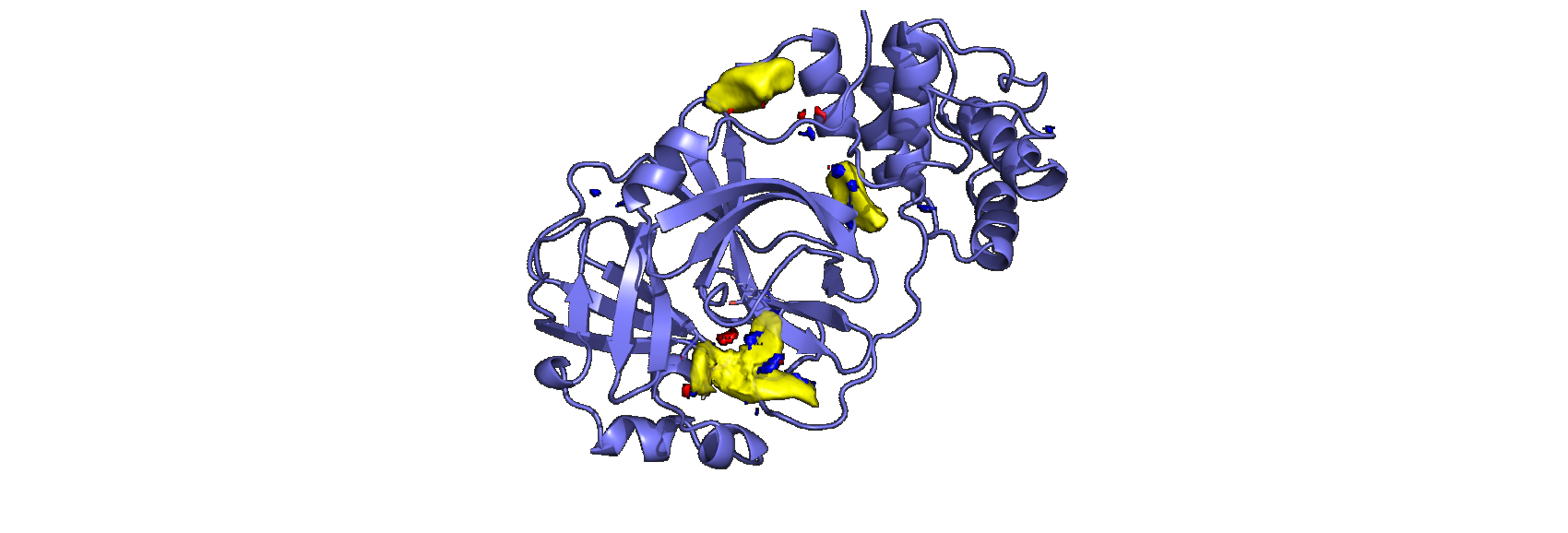
.
We will soon be making the information used to generate these maps available, so we can generate a useful pharmacophore for binding to this key protein.
Our scientists in-house at CCDC are also undertaking a review of the structural information available to understand the best protocols to use in assessing inhibitor designs. We are using our cross-cavity comparison tools to search for related pockets in other targets, and plan to use CSD-CrossMiner to propose alternative design choices as part of the Postera COVID-19 Moonshot effort here.
.
Getting ready for docking
The University of Cambridge HPC centre has made CPU available for use by our collaborators interested in modelling COVID-19.
CCDC is working with the HPC team to get the GOLD (Genetic Optimisation for Ligand Docking) docking software available on their cluster so that collaborators can take advantage of this, again to help as part of the Postera COVID-19 Moonshot effort.
.
 The University of Cambridge HPC centre
The University of Cambridge HPC centre
.
If you’d like to take advantage of these in COVID-19 driven research please contact us at
.
Can we help you?
We are keen to make software and services available to researchers undertaking COVID-19 research. If you need access to our software for this, please let us know on