CSD in Action: Predicting the Formation of Drug Solvent Adducts by Computational Crystal Structure Prediction
In this blog we discuss work that used Cambridge Structural Database (CSD) and CCDC software to support a study related to the potential use of computational crystal structure prediction (CSP) to predict the formation of solvated forms of drug candidates. This is part of our series highlighting applications of the Cambridge Crystallographic Data Centre (CCDC) data and cheminformatics tools by scientists in industry and academia.
Dr Luca Iuzzolino of MRL, Merck & Co., Inc. reports in the journal Crystal Growth & Design (2021, 21, 8, 4362–4371), an approach to using cheminformatics combined with stability calculations to understand the predictability of the formation of drug solvent adducts, mitigating the risk of late form changes in drug development and the potential catastrophic effects these can bring.
Why
Dr Iuzzolino’s role at Merck involves leveraging data to address pharmaceutical development challenges, including solid form analysis, crystal structure prediction, and property prediction.
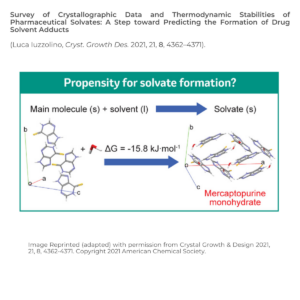
As molecular properties are solid form dependent, solid form selection is a key step in drug development. The same candidate packed in different ways in the solid state can have remarkably different thermodynamic, kinetic, surface, and mechanical properties. These changes impact drug solubility, processing, manufacturing, and stability.
Solid form screening is a key component in drug development as late form changes, such as solvates/hydrates, can have detrimental impacts.
As drug molecules are exposed to solvents in processing, and solvates can display different properties, drug-solvent adducts need to be identified during solid form screening and selection. It would be beneficial to predict whether there is a risk of solvate formation in a drug candidate earlier, to avoid unexpected delays or issues in its development.
What is CSP and how is it used in the pharmaceutical industry?
Crystal Structure Prediction, CSP, is a computational workflow for generating molecular crystal structures from a 2D chemical diagram. It aims to predict the most likely forms to be observed experimentally as well as the relative stability of different possible forms.
Used extensively and routinely in the pharmaceutical industry for de-risking neat (non-solvent) crystal structures, CSP is less used for solvate prediction because:
Challenge 1: Large search space—there are many potential solvates and hydrates with different stoichiometries making CSP of these computationally intensive.
Challenge 2: It is unclear to what extent solvate formation is thermodynamically driven.
How did the Cambridge Structural Database (CSD) and CCDC help to overcome these obstacles?
“CSD data and software tools, with a special mention to the CSD Python API, were instrumental for this work as they allowed us to retrieve, narrow down and analyze a set of solvates of drug-like molecules and extract meaningful statistics from it. The ability to obtain streamlined information on the number of molecules in the asymmetric unit, compound formulae and chirality of these crystal structures was what enabled the analysis of the data. Furthermore, the Crystal Packing Similarity tool, the possibility of adding missing hydrogens and to obtain information on disorder was fundamental to perform calculations to determine the stability of solvates relative to their single component constituents,”
Dr Luca Iuzzolino, MRL, Merck & Co.
The use of the Cambridge Structural Database (CSD) and of the CCDC software to analyse the data provided an invaluable resource to enable an understanding of the applicability of CSP for predicting solvate formation of drug candidates explained Dr Iuzzolino.
CCDC software used included:
Crystal Packing Similarity tool—to compare quantum-mechanically optimized and experimental crystal structures.
CSD Python API—to construct and combine detailed search and analysis functionality to produce tailored scripting workflows.
ConQuest—for advanced 3D searching of structures in the CSD.
“The use of the Cambridge Structural Database (CSD) and of the CCDC software to analyse the data provided an invaluable resource to enable an understanding of the applicability of CSP for predicting solvate formation of drug candidates,” Dr Iuzzolino.
Overcoming Challenge 1: large search space.
A statistical analysis of drug-like solvates present in the CSD was performed to quantify how many could be realistically analysed and predicted by CSP. This gave a solvate data set of 12,000 solvates, of which 7,000 were hydrates and 5,000 non-hydrates.
Overcoming Challenge 2: to what extent is solvate formation thermodynamically driven?
Dr Iuzzolino calculated the relative stabilities of the solvates of drug molecules from non-disordered crystal structures from the CSD drug subset using using quantum mechanical calculations.
In both challenges Dr Iuzzolino used CCDC software to collect and clean a relevant data set from the CSD, then analyse and/or perform calculations on this data.
Conclusion
This work showed that 50-70% of solvates are accessible by CSP. As solvate formation is mostly thermodynamically driven, CSP can predict the stability of solvates relative to the free forms.
“CSP can de-risk the potentially detrimental late formation of solvated forms of drug molecules,” concluded Dr Iuzzolino.
Further work now focuses on whether CSP can be used to predict solvate non-formation.
Next steps
Read the full paper – Survey of Crystallographic Data and Thermodynamic Stabilities of Pharmaceutical Solvates: A Step toward Predicting the Formation of Drug Solvent Adducts (Luca Iuzzolino, Cryst. Growth Des. 2021, 21, 8, 4362–4371) at https://pubs.acs.org/doi/10.1021/acs.cgd.1c00265.
Dr Iuzzolino gave a presentation on this work, watch the recording here.
Find out more about the CSD and the software used in this research.
Read more case studies, including from scientists at Novartis, Bristol-Myers Squibb, GSK and more.