CSD in Action: Training a Machine Learning Model to Predict MOF Pore Accessibility with 80% Certainty
Here we highlight recent work that used metal-organic framework structures in the Cambridge Structural Database (CSD) to train a machine learning model to predict guest accessibility with over 80% certainty. Part of our series highlighting the use of the CSD by scientists around the world.
Summary
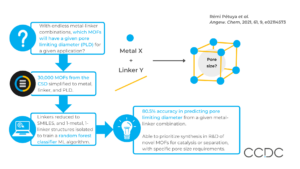
Metal-organic frameworks (MOFs) can be designed to suit specific applications by choosing the metal and linker components. However, the structural, physical, and chemical properties of a MOF, including whether pores are accessible, may not be known until after synthesis. Here, scientists at the University of Liverpool developed a machine learning tool to predict the porosity of MOFs before synthesis, based only on knowledge of the metal and linker.
Why predict MOF pore size?
There is growing interest in diverse applications of MOFs, from gas adsorption, catalysis, drug delivery and more. However, the possible combinations of metals and linkers is endless, so being able to predict which ones will produce the desired properties before synthesis would save time and resources in the lab.
The accessibility of pores is of key importance in catalysis and separation applications of MOFs. It can be assessed by measuring the pore limiting diameter (PLD), defined as the largest free sphere which can diffuse through the structure or, equivalently, the minimum restricting aperture along the diffusion path.
By developing a tool to predict PLD based on accessible descriptors, such as metal and linker molecule, scientists can prioritize their experimental work earlier in the process.
How CSD data trained the machine learning model
First, over 30,000 MOF crystal structures from the CSD were processed. Non-bonded species were identified and removed using the CSD Python API. The metal, linker, and PLD were then identified by a standard simplification algorithm.
The isolated linker structures were reduced to SMILES strings, which were used to calculate 2D descriptors and 3D conformations of the linkers. This avoided data leakage; knowledge of the linker conformations from the CSD being available to the machine learning algorithm.
Structures containing only one metal and one linker were isolated, and used to train the machine learning models to predict MOF porosity.
Outcome: 80.5% accuracy in predicting MOF pore size
With an 80/20 train/test split of the data, a random forest classifier gave 80.5% accuracy when predicting if a given linker-metal combination would produce a MOF with a given pore size.
Furthermore, the group applied sequential learning approaches to further machine learning models to predict if a MOF would have small, medium, or large pore size based on specific angstrom limits. These produced 76% and 68% accuracy, having been trained on smaller subsets of the data.
From the author:
 “The Cambridge Structural Database provides access to about 100,000 MOF structures which is a great resource for design of these materials. For 3D MOF, our work derives additional information from this dataset, defining the constituent linkers and metal atoms in the MOF structures directly from the reference repository of experimentally determined structures.
“The Cambridge Structural Database provides access to about 100,000 MOF structures which is a great resource for design of these materials. For 3D MOF, our work derives additional information from this dataset, defining the constituent linkers and metal atoms in the MOF structures directly from the reference repository of experimentally determined structures.
Accessing CSD structures information through its Python API, such as organic-inorganic bonds, has been a corner stone of the decomposition approach implemented in the database mining.”
Dr Rémi Pétuya
Learn more
Read the full paper in Angewandte Chemie here: https://doi.org/10.1002/anie.202114573
See other examples of the CSD in use in the literature in our collection of case studies here.