CSD-Materials in Action: Molecular Complementarity component reduces required benchtop experiments by 24%
Here we highlight a paper by authors at University of Limerick who used CCDC’s Molecular Complementarity component in Mercury to predict co-crystalization—finding a 24% reduction in the benchtop experiments required to identify the same co-crystals. This is part of our series highlighting examples of the Cambridge Crystallographic Data Centre (CCDC) tools in action by scientists around the world.
Summary
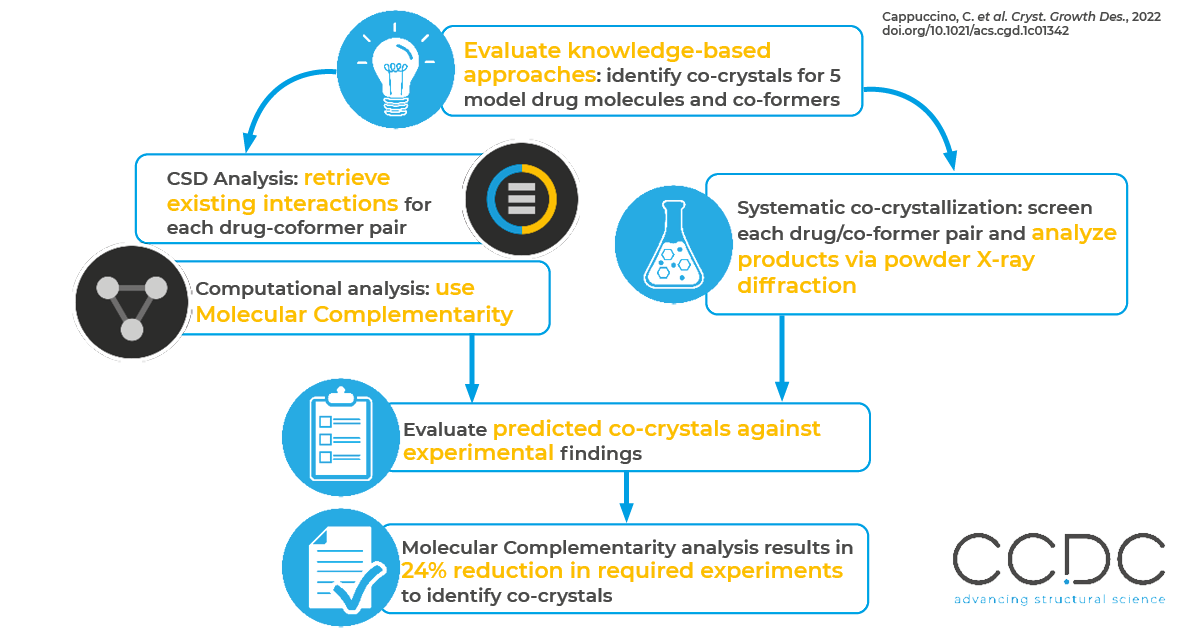
Early-phase drug development requires extensive screening for crystalline forms of potential new active pharmaceutical ingredients (APIs). In this work, researchers evaluate the advantages of knowledge-based approaches in identifying co-crystals for new APIs. They compare two types of analysis to the results of an experimental, systematic crystallization screening:
- An extensive analysis of the Cambridge Structural Database (CSD) to retrieve existing examples of interactions between the functional groups of each drug-coformer pair.
- A computational analysis with Molecular Complementarity.
The Molecular Complementarity component takes into account both molecular energetic and geometrical factors. Of the two approaches, it performed best.
- Molecular Complementarity enabled the identification of all of the observed co-crystals with a 24% reduction of the required experimental attempts.
Why
In early-phase drug discovery, researchers attempt to identify polymorphs with suitable stability and beneficial features, like good solubility. Co-crystallization of multiple molecular components can provide additional positive characteristics, improving the bioavailability of potential APIs. However, testing every potential drug-coformer pair at the benchtop in the hopes of finding an advantageous co-crystal can prove costly. A good computational method can help narrow down the number of required experiments, saving time and resources.
How
First, the researchers selected five drug molecules and five model co-formers for the study. To identify the drug molecules, they searched the CSD for single-component structures associated with the term “drug bank” that contain 20 or fewer carbons—a common threshold for small molecules. To identify the co-formers, they sought compounds with common functional groups with both hydrogen-bond donors and acceptors that are often used in crystal engineering.
The researchers then predicted likely co-crystals with their two knowledge-based methods: an extensive analysis of the CSD and computational analysis with Molecular Complementarity. For the Molecular Complementarity approach, they used two separate thresholds in the software—a 50% likelihood of co-crystallization and a 33% likelihood of co-crystallization.
To evaluate their predictions, the researchers completed a systematic co-crystallization screening of each drug/co-former pair in a 1:1 ratio. They analyzed the products of each crystalization by powder X-ray diffraction. With the Molecular Complementarity approach (using a threshold of 33% likelihood), they found they could predict 100% of the new crystals while performing only 76% of the experiments—for a 24% reduction in the needed experiments.
Learn more
Read the full paper: How Many Cocrystals Are We Missing? Assessing Two Crystal Engineering Approaches to Pharmaceutical Cocrystal Screening. Cryst. Growth Des., 2022.
View more examples of scientists all over the world using CCDC software to advance structural science research.
Tags
CSD (154)
CSD-Materials (29)
Drug Development (46)
Drug Discovery (62)
Mercury (26)
Molecular Complementarity Tool (3)
Pharmaceutical Discovery (37)
Pharmaceuticals (46)
Tools in Action (37)